

Translate this page into:
Giant cell transformation of podocytes: A unique histological feature associated with cystinosis
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Fanconi's syndrome is an unusual cause of renal insufficiency in pediatric patients. Infantile cystinosis is one of the identifiable and treatable etiologies of Fanconi's syndrome. Early diagnosis of cystinosis permits institution of specific therapy with cysteamine. A 3-year-old girl presented with failure to thrive, polyuria, and polydipsia. She was found to have renal tubular defect with renal dysfunction and bilateral small contracted kidneys. A renal biopsy revealed extensive giant cell transformation of podocytes in the glomeruli with focal tubular atrophy and dilatation. However, no crystals were identified. Subsequent ophthalmoscopic examination revealed fine cystine crystals in the cornea and a diagnosis of cystinosis causing Fanconi's syndrome was made. Polykaryocytic transformation of visceral epithelial cells is an important diagnostic clue of nephropathic cystinosis and should be carefully looked for in renal biopsy from a child with Fanconi's syndrome and renal insufficiency.
Keywords
Podocytes
giant cell transformation
cystinosis

Introduction
Fanconi's syndrome is a defect of proximal tubular transport, comprising aminoaciduria, glucosuria, and phosphaturia. It may occur as a idiopathic disease or secondary to a heritable recognizable metabolic disease. Of the latter, cystinosis is the most frequent cause in children.[1] Diagnosis of cystinosis may be made by demonstrating increased concentration of cystine in peripheral leucocytes or cystine crystals in cornea or tissue biopsy by polarization microscopy. However, the crystals are usually dissolved during tissue processing.[1] Apart from the detection of cystine crystals in renal biopsy, presence of multinucleated podocytes is helpful in the diagnosis of this condition.[2] Due to the rarity of this disease, the significance of this subtle finding in renal biopsy is not widely appreciated.
We describe the case of a young girl presenting with renal insufficiency and proximal tubular defects. Renal biopsy showed polykaryocyte visceral epithelial cells and subsequent evaluation confirmed the diagnosis of cystinosis. This rare entity and unusual histological feature is briefly discussed.
Case Report
A 3-year-old female child was referred to our institution for failure to thrive, polyuria, polydipsia, and salt craving. On investigations, she was found to have anemia and renal insufficiency (serum urea 62 mg/dL, serum creatinine 2.1 mg/dL). Urinalysis revealed phosphaturia, increased bicarbonate excretion, glucosuria, and aminoaciduria. Examination of 24-h urine sample showed protein excretion of 1.2 g. Serological investigations (hepatitis B, hepatitis C, human immunodeficiency virus) were negative and serum complement (C3) was within reference ranges. Ultrasonography of the abdomen showed small contracted kidneys. With a clinical diagnosis of proximal renal tubular defect, a renal biopsy was performed.
The renal biopsy showed an adequate core of renal cortical tissue. The glomeruli showed focal mesangial matrix expansion without significant increase in cellularity [Figure 1a]. A striking abnormality was the giant cell transformation of visceral epithelial cells involving almost all the glomeruli [figure 1b and c]. There was no crescent formation or features of collapsing glomerulopathy. Focal tubular dilatation and atrophy with interstitial fibrosis was noted [Figure 1d]. Arterioles showed medial thickening and hyaline arteriosclerosis. The features were suggestive of a metabolic disease, most likely cystinosis. However, no cystine crystals were noted on polarizing microscopy.
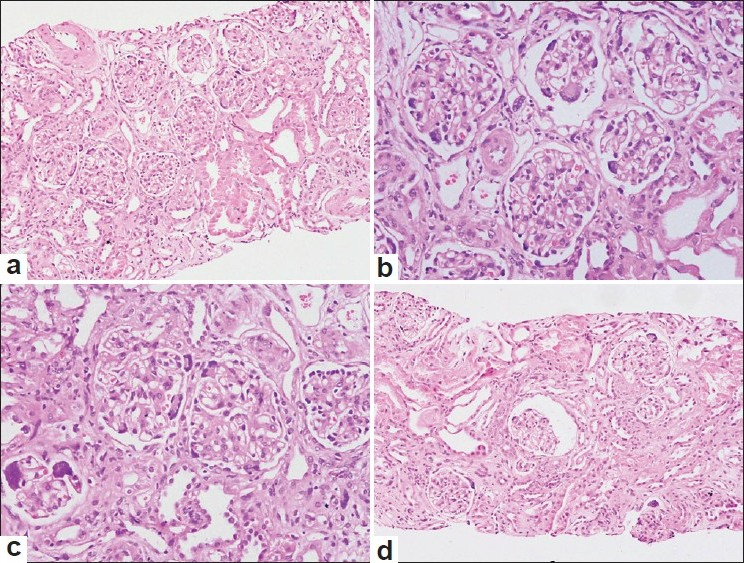
- Low-power photomicrograph of kidney biopsy showing giant cells in the glomeruli (a, H and E × 100). Higher power view demonstrates the multinucleated podocytes in most of the glomeruli (b and c, H and E × 200). An area of tubular dilatation and focal tubular atrophy is also noted (d, H and E × 100)
Immunofluorescence was negative for all immunoglobulins and C3c. Electron microscopy failed to reveal crystalline structures in the epithelial cells. Visceral epithelial cells (podocytes) showed focal degenerative changes with accumulation of phagolysosomes.
Subsequent to the renal biopsy findings, a slit-lamp ophthalmoscopic examination was performed. Fine cystine crystals were observed in the cornea [Figure 2]. Leucocyte assay for cystine could not be performed. A final diagnosis of Fanconi's syndrome caused by cystinosis was made.

- Slit-lamp examination showing fine cystine crystals in the cornea
Discussion
Cystinosis is a rare autosomal recessive lysosomal transport disorder occurring at an estimated incidence of 1 in 100,000 to 200,000 live births.[3] It is caused by the deficiency of cystinosin, a lysosomal membrane protein encoded by a gene, CTNS, mapped to chromosome 17p13.[4] Due to impaired transport of cystine, crystal formation occurs in the reticuloendothelial system and selected epithelia like cornea, thyroid, and renal tubules. Hence, severely affected patients have visual impairment, hypothyroidism, and Fanconi's syndrome.[5] Cystinosis is classified into three forms: (1) infantile nephropathic cystinosis (INC) with rapid progression to end-stage renal disease; (2) juvenile or late-onset cystinosis with slow progression without Fanconi's syndrome; and (3) adult non-nephropathic cystinosis characterized by ocular involvement.[6] INC presents with failure to thrive and complete Fanconi's syndrome. Occasionally, infants present with Bartter's syndrome, diabetes insipidus, and renal insufficiency with proteinuria or photophobia. In such patients, true Fanconi syndrome is recognized only later in the course of disease.[5]
Cystine crystals, though present in substantial quantities in renal biopsy in children with INC, is usually not demonstrable in formalin-fixed and paraffin-embedded tissue sections due to the solubility of cystine crystals in aqueous solutions. In a child with clinical suspicion of cystinosis, the renal biopsy should be fixed in 100% alcohol and directly processed into paraffin to demonstrate the birefringent hexagonal, rhombohedral, or polymorphous cystine crystals on polarization microscopy.[6] A “swan-neck” deformity of proximal tubules has been described; however, it is not specific for cystinosis.[7] Other features like nonspecific chronic damage with tubular atrophy and interstitial fibrosis are frequently seen in biopsy specimens. Patients with heavy proteinuria may show focal segmental glomerulosclerosis (FSGS), with cystinosis as a secondary cause of FSGS.[6]
A histological finding that helps in diagnosis, is the presence of multinucleated podocytes. Multinucleation may also be observed in tubular epithelial and interstitial cells.[268] This feature is considered to be relatively specific due to the rarity of this alteration in other renal diseases, as identified by Nagata et al. in their study.[9] Multinucleated giant cells of macrophage origin may be seen in necrotizing glomerular diseases especially antiglomerular basement membrane disease. The presence of tuft necrosis with fibrin and cellular crescents helps in differentiating such multinucleation from polykaryocytes of cystinosis.[10] The pathogenesis of podocyte multinucleation in cystinosis is not known, since podocytes are considered to be terminally differentiated cells. Experimental studies have shown proliferative capabilities, especially nuclear division, in podocytes after treatment with basic fibroblast or platelet-derived growth factor.[1112] In humans, dedifferentiation of podocytes with expression of proliferation marker, Ki-67, has been documented in collapsing glomerulopathy.[13] It has been hypothesized that podocytes undergo nuclear division in cystinosis, though cell division does not ensue leading to multinucleated podocytes.[6] In the present case, multinucleation of podocytes involved majority of the glomeruli and there was no morphologic evidence of collapsing glomerulopathy.
The optimal therapeutic modality for cystinosis, diagnosed at an early age, is oral cysteamine, which reacts with cystine to form complexes capable of transport out of the lysosomes. Dialysis and renal transplantation are required for patients presenting in renal failure.[6]
This case is being reported due to the rarity of this condition as one of the detectable causes of Fanconi's syndrome. Multinucleation of glomerular visceral epithelial cells is a unique alteration helpful in early diagnosis of this rare heritable condition, the definite diagnosis of which requires determination of cystine levels in peripheral lymphocytes or the presence of characteristic crystals in cornea.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Renal diseases caused by familial metabolic and hematologic diseases, in Heptinstall's pathology of the kidney. (6th edn). Philadelphia: Lippincott Williams and Wilkins; 2007. p. :1220-21.
- [Google Scholar]
- Polykaryocytes of the visceral glomerular epithelium in cystinosis with description of an unusual clinical variant. John Hopkins Med J. 1971;129:83-99.
- [Google Scholar]
- A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319-24.
- [Google Scholar]
- Multinucleated podocytes in a child with nephrotic syndrome and Fanconi's syndrome: a unique clue to the diagnosis. Am J Kidney Dis. 1999;34:966-71.
- [Google Scholar]
- Adolescent cystinosis: comparison with the infantile and adult forms. Pediatrics. 1971;47:979-88.
- [Google Scholar]
- Adolescent cystinosis, presenting with proteinuria. Int J Pediatr Nephrol. 1983;4:79-81.
- [Google Scholar]
- Mitosis and the presence of binucleate cells among glomerular podocytes in diseases human kidneys. Nephron. 1995;70:68-71.
- [Google Scholar]
- Pathogenic significance of interleukin-6 in a patient with anti-glomerular basement membrane antibody-induced glomerulonephritis with multinucleated giant cells. Am J Kidney Dis. 1995;26:72-9.
- [Google Scholar]
- Long-term treatment of rats with FGF-2 results in focal segmental glomerulosclerosis. Kidney Int. 1995;48:1435-50.
- [Google Scholar]
- Visceral glomerular epithelial cells can proliferate in vivo and synthesize platelet-derived growth factor B-chain. Am J Pathol. 1993;142:637-50.
- [Google Scholar]
- The dysregulated podocyte phenotype: A novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 1999;10:51-61.
- [Google Scholar]




