

Translate this page into:
Neonatal Bartter syndrome associated with ileal atresia and cystic fibrosis
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
A rare case of neonatal Bartter syndrome presenting with severe hyperkalemia is reported in a preterm child born to consanguineous parents. This child also had ileal atresia, and meconium plugs were found at laparotomy. The diagnosis of cystic fibrosis was subsequently made on genetic testing. Despite full intensive care management and surgical interventions, he died of respiratory failure after 70 days. This is the first reported case of such conglomeration of pathologies in a newborn child. Second, in highlighting this case we want clinicians to be aware that a subtype of neonatal Bartter syndrome can present with initial hyperkalemia so that an erroneous diagnosis of pseudohypoaldosteronism is not made when this is seen in combination with hyperkalemia and hyperrenin hyperaldosteronism.
Keywords
Cystic fibrosis
ileal atresia
neonatal bartter syndrome
pseudo-hypoaldosteronism

Introduction
The term Bartter syndrome (BS) refers to a group of rare congenital renal tubular disorders characterized by hypokalemia, hypochloridemia, metabolic alkalosis, and hyper-reninemia without hypertension.[1] The primary defect relates to abnormal salt reabsorption in the thick ascending loop of Henle (TALH) and the distal convulated tubule (DCT). BS is inherited in an autosomal recessive fashion and five genetic subtypes have been described.[2–4] The neonatal (or antenatal) form is characterized by polyhydramnios, preterm rupture of membranes (pPROM), and preterm labor leading to prematurity; and two subtypes have been described. In the immediate newborn period, there is associated polyuria with resultant severe dehydration, electrolyte imbalance (including hypokalemia), and metabolic alkalosis. They also experience failure to thrive and have dysmorphic facies.[5]
One subtype of neonatal BS presenting with hyperkalemia have been reported recently.[6–8] This presence of hyperkalemia coupled with hypokalaemia and hyper-renin hyperaldosteronism can easily be “misdiagnosed” as pseudo-hypoaldosteronism type 1. We are reporting a case of neonatal BS that was suspected antenatally because of family history and pregnancy complicated by polyhydramnios. This male baby was born at 29 weeks gestation and developed hyperkalemia in the immediate newborn period. He was subsequently found to have ileal atresia and cystic fibrosis. Clinicians should be aware of this unusual presentation of neonatal BS to forestall erroneous diagnosis and delay of appropriate investigations and interventions.
Case Report
A male infant was born to consanguineous healthy parents at 29 + 6 week of gestation. The mother has had three previous pregnancies and the first two were complicated by polyhydramnios, leading to preterm deliveries at 26 and 32 weeks, respectively. The first child died in the early neonatal period of an unexplained etiology whereas the second was diagnosed as having pseudohypoaldosteronism. The correct diagnosis of BS was subsequently made when this child developed nephrocalcinosis at the age of 4 years. Both parents are carriers of the KCNJ1 gene. Their third child is healthy and unaffected.
This pregnancy was complicated by polyhydramnios and gestational diabetes. Antenatal genetic testing was offered, but was declined. Mother developed premature rupture of membrane at 26 weeks gestation and received antenatal steroids. She was delivered by emergency caesarean section because of abnormal fetal monitoring, but Apgar scores were normal. He weighed 1.33 kg (25th-50th centiles) and his head circumference was 27 cm (25th centiles). Clinical examinations showed no facial dysmorphisms.
He remained clinically stable in the 1st 24 h of life receiving routine care. However, on day 2 he developed hyperkalemia, hypocalcaemia, hyponatremia, and polyuria. The maximum serum potassium level was 8.5 mmol/l (ref 3.5-4.5 mmol/l). This proved difficult to control needing bicarbonate and calcium gluconate infusions, salbutamol nebulization, and calcium resonium enema. There was no associated cardiac arrhythmia. The lowest serum sodium was 120 mmol/l on day 6, and he required sodium supplementation to a maximum requirement of 28 mmol/kg/day on day 8 of life. The polyuria led to significant weight loss and dehydration necessitating liberal fluid replacement to a maximum of 240 ml/kg/day on day 8 of his life. His maximum urine output measured 155 ml/kg/day on day 7. He also needed calcium and magnesium supplementation to maintain normal electrolyte balance. Urinary electrolyte analysis showed extensive sodium, potassium, and chloride loss and high urinary calcium/creatinine ratio. The initial hyperkalemia gave way to hypokalemia and metabolic alkalosis during the 2nd week of life. Thereafter, he required potassium replacement therapy to a maximum of 6 mmol/kg/day.
In view of his presentation and family history a diagnosis of neonatal BS was considered. The Genetic and Nephrology teams reviewed him and genetic testing showed KCJN1 gene mutation; confirming the diagnosis of Neonatal BS subtype 2. He also had a normal male karyotype and very high serum aldosterone level (41,000 pmol/l, ref 111-862 pmol/l). The serum renin, 17-hydroxyprogesterone, and angiotensin levels were all within normal limit.
Further progress
He continued to need persistently high fluid requirement (190-220 ml/kg/day) as parenteral nutrition (PN) with concomitant high urine output of (7-10 ml/kg/day). Following stabilization of his fluid and electrolyte requirements, enteral feeding was started using expressed maternal breast milk (EBM), on day 6 of life. He developed abdominal distension and bilious aspirates on day 12 which was treated as necrotizing enterocolitis (NEC). However, over the next 48 hours radiological evidence of intestinal obstruction became evident. On day 14 of life, he underwent laparotomy where ileal atresia, microcolon, and meconium plugs were found. Resection of the atretic segment with end-to-end anastamosis was performed. Histopathology examination of the ileal specimen was suggestive of cystic fibrosis and genetic testing for cystic fibrosis was undertaken. Thereafter, enteral feeds were reintroduced and treatment with N-acetyl cysteine tried to help manage the meconium plugs. He showed poor response to this treatment and developed further episode of abdominal distension, increasing residuals and infrequent bowel opening. This second episode of abdominal concerns coincided with respiratory deterioration necessitating a second laparatomy on day 36. There was extensive intraperitoneal adhesion and ileostomy with mucus fistula was fashioned just distal to the earlier site of ileal anastamosis. Enteral feeds was reintroduced and slowly advanced to full feed on day 56. He however, continued to have poor weight gain and renal ultrasonogram done on day 34 showed nephrocalcinosis [Figure 1].
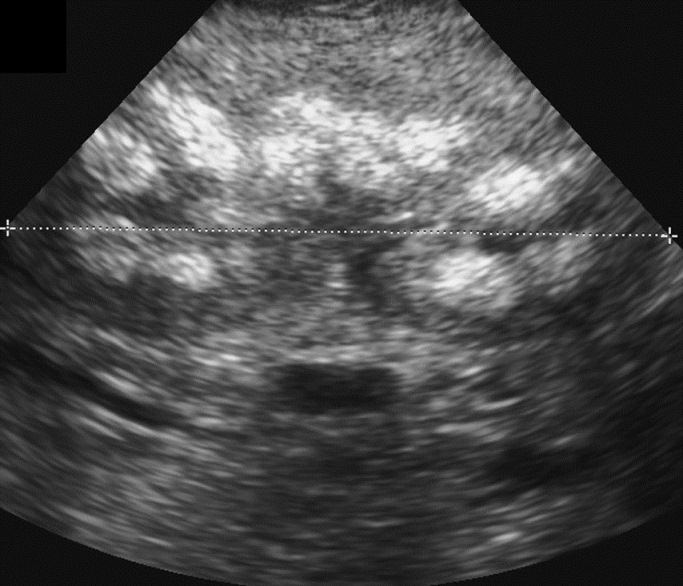
- Renal ultrasonogram of the right kidney showing highly echogenic renal medulla and no acoustic shadows: features are more than expected from prematurity but suggestive of nephrocalcinosis. The left kidney was equally affected
His respiratory functions deteriorated following surgery leading to escalating support – both conventional and high frequency oscillation ventilation (HFOV). His chest radiograph also worsened, showing segmental collapse, cystic changes and areas of localized hyperinflation. He was treated with antimicrobial agents and needed a ten day course of postnatal steroids to help wean him from the ventilator on day 42 unto continuous positive airway pressure (CPAP) support. Genetic testing for cystic fibrosis later confirmed the presence of homozygous deletion of exon 4-10 of the CFTR gene on day 38 of life. He also had high immunoreactive trypsin level (92 ng/ml, ref. <70) from the newborn screening blood test done on day 5 of life.
Following this new diagnosis he was reviewed by the respiratory, gastroenterology, and dietetic teams with introduction of pancreatic enzyme replacement and regular gentle physiotherapy. Despite these interventions, he developed a further episode of respiratory deterioration necessitating a second course of postnatal steroid, nebulisation of DNAse, and exogenous surfactant therapy (used as a mucolytic). He also needed brief period of mechanical ventilation between days 50 and 55 before weaning to CPAP support. Further respiratory deterioration occurred with steroid reduction and Influenza A viral infection; that was detected on PCR testing of a nasopharyngeal aspirate on day 64. In view of his deteriorating lung functions despite aggressive support his management plan was changed to palliative care provision after agreement with the parents, and he died soon after on day 70 of life.
Discussion
Advances in genetic research continue to shed more light to our understanding of BS since the first report in 1962.[1] The primary defect relates to mutations of genes encoding proteins that transport ions across renal cells in the TALH and DCT of the nephrons and five types are described.[9–14] Neonatal BS is caused by types 1 and 2 genetic defects and differs from classic BS because of the age of onset, presence of nephrocalcinosis, and very high urinary loss of sodium, calcium, and chloride.
Prenatal diagnosis can be made by the presence of high chloride content of the amniotic fluid and from mutational analysis of genomic DNA extracted from cultured amniocytes obtained by amniocentesis.[1516] Pregnancy is complicated by polyhydramnios because of fetal polyuria. There is increased risk of preterm delivery; usually between 24 and 30 weeks gestation. In our report, there was polyhydramnios which could have also been worsened by maternal gestational diabetes. This invariably led to preterm labor.
The main therapeutic challenge in the early neonatal period relates to the management of fluid and electrolyte balance. There is polyuria with excessive urinary loss of electrolyte resulting in hyponatraemia, hypokalaemia, metabolic alkalosis and dehydration. Serum calcium and magnesium level may also be low, although hypomagnesaemia is more typical of Gitelman syndrome.[17] Liberal fluid requirement and replacement of lost electrolytes is warranted and fluid requirements up to 500 ml/kg/day has been reported.[18] In Neonatal BS type 2, there is early hyperkalaemia which can be life threatening without prompt treatment. It is therefore important to recognize this variation in presentation so as not to make an erroneous diagnosis of pseudohypoaldosteronism.[6–8] Moreover, careful attention to the monitoring of serum potassium level is needed because the initial hyperkalaemia will usually give way to hypokalaemia, warranting potassium replacement. Treatment with NSAIDs, ACE inhibitors, and selective COX 2 agents has been shown to be effective in controlling the electrolyte loss from the kidney[171920] but their use in preterm infants is usually avoided for the first 6 weeks of life given the associated complications.[21] We also did not treat our patient with NSAID agent at a later age because of his bowel problems. Potassium sparing diuretics can also be used to ameliorate the associated hypokalaemia, but their effect is transient.[22] Classically, the fluid and electrolyte requirement will generally stabilize after the initial upheavals and survivors will continue to require potassium and sodium supplementation long-term and exhibit poor growth. They also develop nephrocalcinosis with time because of high calciuria as in our case.
In this report, the respiratory functions deteriorated markedly following surgery. This phenomenon is well recognized in patients with cystic fibrosis undergoing surgery under general anesthesia and is thought to be secretion related. This problem could be ameliorated by careful perioperative management involving shared care between the surgeon, anesthetist, and neonatologist.[23] We however could not offer such care since cystic fibrosis was only considered following the intra-operative findings. The coexistence of neonatal BS and cystic fibrosis may not be fortuitous given that both are recognized chloride channel disorders.[24] Patients with BS who develop unexplained chronic respiratory symptoms should therefore be investigated for cystic fibrosis. Following mechanical ventilation postsurgery, he developed findings consistent with chronic lung disease of prematurity (CLD). It is likely that the CLD was made worse by the coexistent cystic fibrosis. Unfortunately, he also contracted H. Influenza A virus infection before he could receive vaccination and his respiratory functions deteriorated further.
The management of a preterm infant with neonatal BS, enterostomy, and cystic fibrosis is challenging. This therefore requires multi-disciplinary involvement including the neonatologist, pediatric surgeons, gastroenterology, respiratory physicians, dietetics, nephrology, and genetics. He required prolonged PN and pancreatic enzyme replacement once substantial quantities of enteral milk were tolerated. We used Pancreas Viokase® which contains pancrelipase in pH sensitive, enteric coated microspheres instead of Creon because of its ease of administration through the nasogastric tube. Infants are usually started at a dose of 2000-4000 IU lipase per 120 ml of formula or per breastfeeding session.[25] The dosage was adjusted based on the consistency and volume of stoma losses over every 24 hour period.
Our patient's multiple pathologies and stormy clinical course evoked many ethical questions regarding the rationale for continuation of intensive care support. Long-term prognosis of babies with neonatal BS is always guarded even in the absence of co-pathologies. These ethical issues were discussed with the parents and within the team until there was consensus. Thereafter, his care plan was rightly refocused to palliative care provision.
Conclusion
This report highlights the complexities of managing a rare inherited disorder such as neonatal BS. Clinicians should be aware that type 2 subtype of neonatal BS presents with early hyperkalemia. This knowledge should help prevent the diagnosis of pseudohypoaldosteronism in error. The degree of combination of pathologies in our report is unprecedented and required multi-disciplinary input. Careful ethical considerations should however be given when considering the appropriateness of continuation of intensive care support in similar cases.
Source of Support: Nil
Conflict of Interest: We declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med. 1962;33:81-28.
- [Google Scholar]
- Bartter's and Gitelman's syndromes: From gene to clinic. Nephron Physiol. 2004;96:p65-78.
- [Google Scholar]
- Type IV Bartter syndrome: Report of two new cases. Pediatr Nephrol. 2006;21:766-70.
- [Google Scholar]
- Autosomal dominant hypocalcemia with mild type 5 Bartter syndrome. J Nephrol. 2006;19:525-8.
- [Google Scholar]
- Bartter syndrome in two siblings-antenatal and neonatal observations. Int J Pediat Nephrol. 1985;6:63-70.
- [Google Scholar]
- Neonatal Bartter Syndrome with Early Hyperkalemia: An Unusual Presentation in Twins. Kuwait Med J. 2005;37:130-2.
- [Google Scholar]
- Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome. J Pediatr. 2003;142:318-23.
- [Google Scholar]
- International Collaborative Study Group for Bartter-like Syndromes: Mutations in the gene encoding the inwardly rectifying renal potassium channel, ROMK, cause the antenatal variant of Bartter syndrome: Evidence for genetic heterogeneity. Hum Mol Genet. 1997;6:17-26.
- [Google Scholar]
- Bartter and related syndromes: The puzzle is almost solved. Pediatr Nephrol. 1998;12:315-27.
- [Google Scholar]
- An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol. 2008;4:560-7.
- [Google Scholar]
- Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet. 1996;13:183-8.
- [Google Scholar]
- Genetic heterogeneity of Bartter's syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet. 1996;14:152-6.
- [Google Scholar]
- Molecular basis of DFNB73: Mutations of BSND can cause nonsyndromic deafness or Bartter syndrome. Am J Hum Genet. 2009;85:273-80.
- [Google Scholar]
- Autosomal dominant hypocalcemia with mild type 5 Bartter syndrome. J Nephrol. 2006;19:525-8.
- [Google Scholar]
- Prenatal and postnatal management of hyperprostaglandin E syndrome after genetic diagnosis from amniocytes. Pediatrics. 1999;103:678-83.
- [Google Scholar]
- A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221-35.
- [Google Scholar]
- Role of cyclooxygenase-2 in hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int. 2002;62:253-60.
- [Google Scholar]
- Angiotensin-converting enzyme inhibition as a therapeutic principle in Bartter's syndrome. Eur J Clin Pharmacol. 1991;41:303-5.
- [Google Scholar]
- Neonatal Bartter syndrome–use of indomethacin in the newborn period and prevention of growth failure. Pediatr Nephrol. 1996;10:756-8.
- [Google Scholar]
- Bartter's syndrome-treatment with potassium, spironolactone and ACE-inhibitor. J Intern Med. 1989;225:107-10.
- [Google Scholar]
- The need to avoid general anaesthesia in cystic fibrosis. J R Soc Med. 1986;79(Suppl 12):10-2.
- [Google Scholar]
- Mutations in the human skeletal muscle chloride channel gene (CLCN1) associated with dominant and recessive myotonia congenita. Neurology. 1996;47:993-8.
- [Google Scholar]
- British National Formulary for children 2010-2011




