

Translate this page into:
Distal renal tubular acidosis and amelogenesis imperfecta: A rare association
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Renal tubular acidosis (RTA) is characterized by a normal anion gap with hyperchloremic metabolic acidosis. Primary distal RTA (type I) is the most common RTA in children. Childhood presentation of distal RTA includes vomiting, failure to thrive, metabolic acidosis, and hypokalemia. Amelogenesis imperfecta (AI) represents a condition where the dental enamel and oral tissues are affected in an equal manner resulting in the hypoplastic or hypopigmented teeth. We report a 10-year-old girl, previously asymptomatic presented with the hypokalemic paralysis and on work-up found out to have type I RTA. The discoloration of teeth and enamel was diagnosed as AI.
Keywords
Amelogenesis imperfecta
distal renal tubular acidosis
hypokalemic paralysis

Introduction
Distal renal tubular acidosis (RTA) is caused mostly by impaired H+ secretion and may be genetic or acquired. The development of systemic acidosis diminishes net proximal fluid reabsorption and increases the distal delivery. This volume contraction activates renin angiotensin aldosterone axis and leads to potassium wasting. Amelogenesis imperfecta (AI) represents a group of condition, genomic in origin, which affect the structure and the clinical appearance of enamel of nearly all the teeth and which may be associated with the morphological or biochemical changes elsewhere in the body.[1]
Dental anomalies are rare in RTA, but have been described occasionally in proximal RTA.[23] We hereby present a case of the distal RTA presenting as hypokalemic paralysis in a 10-year-old child with AI, which to the best of the authors’ knowledge, is the first such reported case in the pediatric age group.
Case Report
A 10-year-old female, resident of Tamil Nadu born of second degree consanguineous marriage was admitted with inability to move all 4 limbs since early morning. The weakness had evolved over a period of 20 days initially presenting as calf muscle pain, progressing to difficulty in getting up from bed, and eventually involving all four limbs.
There was no prior history of fever, loose stools, vomiting, trauma or drug intake. No significant past medical history could be elicited except for polyuria and polydipsia for last 1 month. Antenatal, perinatal, and development history was unremarkable. There was no family history of periodic paralysis, cardiac, thyroid, renal or auto immune disorders. On examination, child had flaccid areflexic quadriparesis with power 2/5 in all limbs and no muscle wasting. Her higher mental functions, cranial nerves, sensory system and autonomic system examination were normal. Systemic examination was unremarkable. Her vitals were within normal range. Serum level of potassium was 1.5 meq/l and electrocardiogram showed prolonged QT interval, ST depression, T wave flattening in V1-V4, and the presence of ‘U’ waves. Child was given potassium replacement therapy and recovered from paralysis.
She had severe growth retardation with a weight of 15 kg and height of 110 cm (both below 5th centile). Intraoral examination revealed poor oral hygiene; the dentition was consistent with the chronological age. Enamel was hypoplastic with brownish orange discoloration and irregular mottling of occlusal surface of molar teeth. Based on the history, clinical and radiographic presentation provisional diagnosis of AI, hypoplastic type was made [Figure 1]. Routine lab investigations including renal and liver function tests were within normal range. Serum electrolytes showed hypocalcemia (7.2 mg/dl), hypophosphatemia (2.8 mg/dl) with normal serum magnesium and sodium. Her arterial blood gas revealed normal anion gap metabolic acidosis with respiratory alkalosis [Table 1]. Thyroid and lipid profile were normal. Urine culture and sensitivity showed no growth and urine metabolic screening were negative. There was no glycosuria or proteinuria. The urinary potassium level was 24 meq/l, which indicates renal potassium wasting (exceeding 20 meq/l).
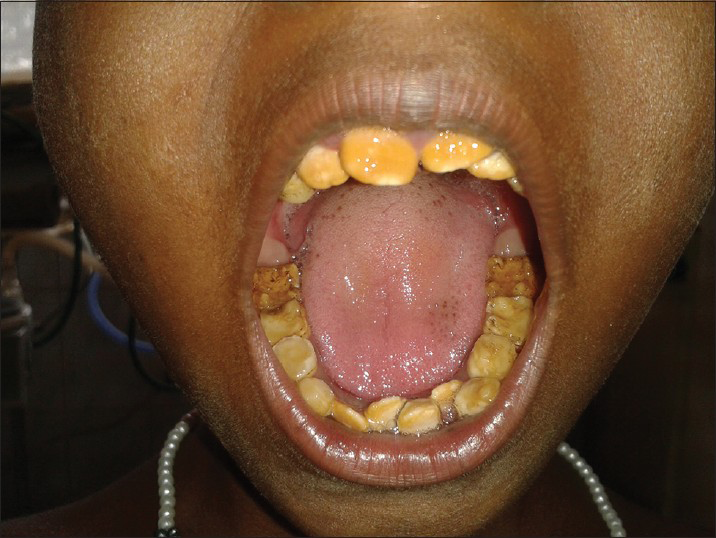
- Photograph of the teeth of the patient showing amelogenesis imperfecta

In view of persisting metabolic acidosis with normal anion gap, hypokalemia, and polyuria (2.5 l/m2/24 h) a possibility of distal RTA was considered, which was confirmed by necessary investigations. Three consecutive early morning urine pH was 7.5 and the urine pH after frusemide challenge was 7.5. Urine anion gap was positive and urinary calcium/creatinine ratio was 2. 24 h urinary calcium was 280 mg (increased), fractional excretion of bicarbonate was 4.5% and urine blood pCO2 difference was 4 mmHg (decreased). The ultrasound scan of the abdomen revealed bilateral renomegaly with increased cortical echoes. There was no evidence of nephrocalcinosis. Skeletal survey did not show any evidence of rickets. She was investigated for secondary causes of RTA and all investigations including her antinuclear antibody and auto immune profile were negative.
The child was managed symptomatically with potassium replacement therapy initially intravenously followed by oral supplementation with correction of acidosis and electrolyte abnormalities during the ICU stay and upon the diagnosis of distal RTA was started on long- term oral potassium citrate supplementation. The patient is currently under follow-up and her weakness and polyuria have improved dramatically.
Discussion
RTA is a condition in which there is a defect in renal excretion of H+ or reabsorption of bicarbonate or both, which occurs in the absence of impairment in glomerular filtration rate.[4] From a pathophysiological perspective RTA is classified as type 1 (distal), type 2 (proximal), and type 4 (hyperkalemic). Primary distal type 1 RTA is the most common type of RTA found in children. It is due to impaired distal acid secretion by alpha intercalated cells that result in an inability to excrete the daily acid load. It can either be inherited or can occur secondarily to a wide variety of clinical disorders.
Primary forms of distal RTA manifest in early infancy with vomiting, polyuria, polydipsia, dehydration crisis, failure to thrive, and growth retardation. Nephrocalcinosis is usually present at the time of diagnosis. Our case is a form of primary distal RTA, which is very characteristic of the pediatric population. Primary distal RTA rarely presents in children with neuromuscular symptoms linked to potassium deficiency including hypokalemic paralysis although this is more common in adults with distal RTA and Sjogren syndrome.[5]
Although, distal as well as proximal RTA can present with periodic paralysis the further work-up carried out in our patient points towards type 1 RTA. The hallmark of distal RTA is the inability to lower the urine pH maximally in the face of moderate to severe systemic acidosis.[4] Acidosis can be provoked by giving oral ammonium chloride (100 mg/kg) or frusemide (40 mg) and monitoring the urine pH over 8 h or 4 h respectively. In distal RTA urine pH should not fall below 5.3.[6] Halperin et al., have shown that an estimation of urine ammonium can be made by determining the urine anion gap.[7] Where elimination of ammonium is abnormally low as happens in patients with distal RTA the urine anion gap remains positive.[8] Coexistence of hypercalciuria and hypocitraturia leads to nephrocalcinosis characteristic of these patients. Hypokalemia of variable degree is frequently found in patients with distal RTA. Its pathogenic mechanism is poorly understood, but it has been related to a low ability of the H+ K+ ATPase pump in the collecting duct.[9]
AI is a group of inherited defects of dental enamel formation characterized by both clinical and genetic heterogeneity. Most of the cases are inherited either as X linked, autosomal dominant, autosomal recessive or sporadically. To date mutations in five genes (AMELX, ENAM, DLX3, KLK4 and MMP20) have been found AI.[10] AI exists in isolation or associated with other abnormalities in syndromes such as cone-rod dystrophy, platyspondyly, nephrocalcinosis, hypothalamo-hypophyseal insufficiency, and Kohlshutter syndrome. The syndrome of AI and nephrocalcinosis, an extremely rare syndrome has been reported in few families under the name of enamel renal syndrome.[1112131415] To our knowledge only two cases of AI and distal RTA have been described.[1617] To the best of the authors’ knowledge this will be the first association of distal RTA with AI reported in the pediatric age group. Most of these cases including our case occurred in consanguineous families and suggest an Autosomal Recessive (AR) pattern of inheritance. The role of dental protein in calcium and phosphate metabolism and renal function needs further research.
Patients with distal RTA usually require only 1-3 meq/kg/day alkali supplementations to correct the metabolic acidosis often correcting kaliuresis and natriuresis as well. Long-term treatment induces catch up growth, arrests progressive nephrocalcinosis and related development of renal failure and bone deformities. Follow-up includes regular assessment for growth and blood levels of electrolytes, pH and bicarbonate. The supportive clinical care needed by patients with AI is substantial both in terms of clinical and emotional demands. The long-term care involves either crowns or adhesive plastic restorations.
Early diagnosis (preferably before 2 years) of distal RTA can minimize the morbidity and improve the growth of these children. Distal RTA can have a wide variety of systemic association and the present association of AI highlights the need for a thorough renal evaluation in children with enamel disorders. The early diagnosis provided by oral symptoms can lead to a better renal prognosis.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Amelogenesis imperfecta: A classification and catalogue for the 21st century. Oral Dis. 2003;9:19-23.
- [Google Scholar]
- Congenital persistent proximal type renal tubular acidosis in two brothers. Acta Paediatr Scand. 1979;68:861-8.0.
- [Google Scholar]
- Dental features in congenital persistent renal tubular acidosis of proximal type. Scand J Dent Res. 1984;92:489-95.
- [Google Scholar]
- Renal involvement and followup of 130 patients with primary Sjögren's syndrome. J Rheumatol. 2008;35:278-84.
- [Google Scholar]
- Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: An alternative to ammonium chloride. Kidney Int. 2007;71:1310-6.
- [Google Scholar]
- Studies on the pathogenesis of type I (distal) renal tubular acidosis as revealed by the urinary PCO2 tensions. J Clin Invest. 1974;53:669-77.
- [Google Scholar]
- Mycophenolate mofetil in steroid/cyclosporine-dependent/resistant nephrotic syndrome. Pediatr Nephrol. 2005;20:914-9.
- [Google Scholar]
- Interventions for idiopathic steroid-resistant nephrotic syndrome in children. Cochrane Database Syst Rev. 2010;11:CD003594.
- [Google Scholar]
- Genes and related proteins involved in amelogenesis imperfecta. J Dent Res. 2005;84:1117-26.
- [Google Scholar]
- Syndrome of amelogenesis imperfecta, nephrocalcinosis, impaired renal concentration, and possible abnormality of calcium metabolism. Am J Med Genet. 1985;20:233-43.
- [Google Scholar]
- Amelogenesis imperfecta and nephrocalcinosis syndrome. Case studies of clinical features and ultrastructure of tooth enamel in two siblings. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;79:583-92.
- [Google Scholar]
- Amelogenesis imperfecta, nephrocalcinosis, and hypocalciuria syndrome in two siblings from a large family with consanguineous parents. Nephrol Dial Transplant. 1998;13:3193-6.
- [Google Scholar]
- Amelogenesis imperfecta and nephrocalcinosis: A new case of this rare syndrome. J Clin Pediatr Dent. 2003;27:171-5.
- [Google Scholar]
- Amelogenesis imperfecta and distal renal tubular acidosis presenting as hypokalemic periodic paralysis. J Assoc Physicians India. 1999;47:1205-6.
- [Google Scholar]
- Amelogenesis imperfecta with renal disease: A report of two cases. J Oral Pathol Med. 2007;36:625-8.
- [Google Scholar]




