

Translate this page into:
Karyomegalic interstitial nephritis with focal segmental glomerulosclerosis: A rare association
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Karyomegalic interstitial nephritis (KIN) is a rare form of, progressive chronic interstitial nephritis. We present a case of KIN in a child, who was also found to have nephrotic syndrome because of focal segmental glomerulosclerosis on renal biopsy. To our knowledge, this is the first case of KIN associated with glomerulopathy.
Keywords
Focal segmental glomerulosclerosis
interstitial nephritis
karyomegaly
nephrotic syndrome

Introduction
Karyomegalic interstitial nephritis (KIN) was first described in 1974.[1] A detailed description of patients with history of recurrent respiratory infections and progressive renal failure was given 1979.[2] The prevalence of this disease is less than 1%.[3] The pathogenesis of KIN is unclear. The disease has no known treatment. The case under discussion has various rare features like young age of the patient, associated glomerulopathy causing nephrotic syndrome and normal renal function, which is showing gradual deterioration. Normal renal function at the time of diagnosis is known and in one large series published by Bhandari et al., one out of the six cases presented with microhematuria and normal renal function.
Case Report
An 8-year-old male child presented with a history of facial puffiness and edema of one year duration. He was diagnosed to have nephrotic syndrome and was treated by a local physician with steroids. He did not improve, however, and was referred to this hospital for further management.
There was no family history of kidney disease. He is the first child of non-consanguineous marriage. There was no history of drug ingestion including exposure to mycotoxins or other herbal medicines. No history of recurrent respiratory symptoms. On examination, patient had stunted growth with moon facies, secondary to steroids and had clinical features of rickets.
His investigations revealed hemoglobin of 10 g/dl, blood urea 33 mg/dl and serum creatinine 0.7 mg/dl. His liver functions revealed hypoalbuminemia of 2.6 g/dl. His liver enzymes were within normal limits. He was human immunodeficiency virus non-reactive and hepatitis B surface antigen and hepatitis C virus and 24 h urine protein was 3565 mg/dl. Complete urine examination showed 2+ albumin, 4-6 red blood cells and granular casts. Ultrasound revealed normal sized kidneys with normal echotexture.
He was started on cyclophosphamide. However, there was no response even after 6 months. Hence a kidney biopsy was done. Biopsy showed 23 glomeruli, four of whom showed segmental sclerosis [Figure 1]. Basement membrane and cellularity were normal in the uninvolved glomeruli. Tubular epithelial cells revealed changes diffusely. The lining cells in many cells revealed enlarged nuclei with lobulated appearance. These cells are larger than the normal tubular epithelial cells [Figure 2]. There was chromatin clumping giving a hyperchromatic appearance. Mitoses were sparse. There is atrophy of the tubules with thickening of tubular basement membrane. There is interstitial fibrosis and interstitial mild lymphomononuclear infiltration [Figure 3].
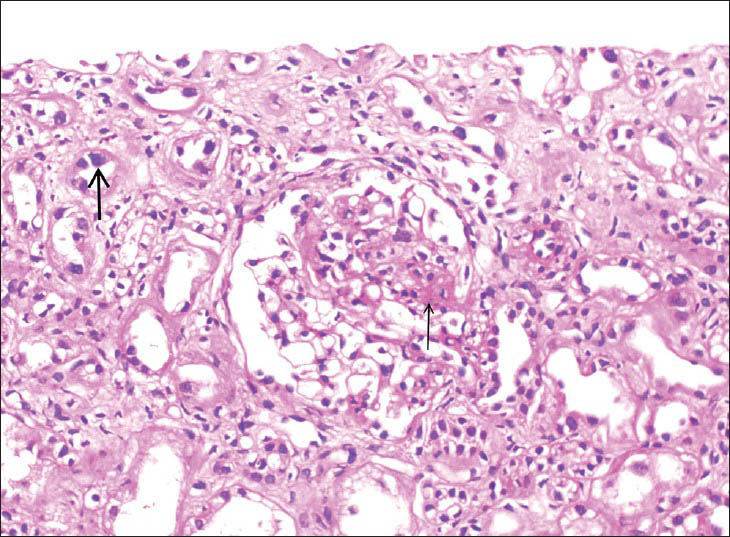
- Segmental glomerulosclerosis (thin arrow) and karyomegaly in tubular cells (thick arrow) (H and E, ×400)
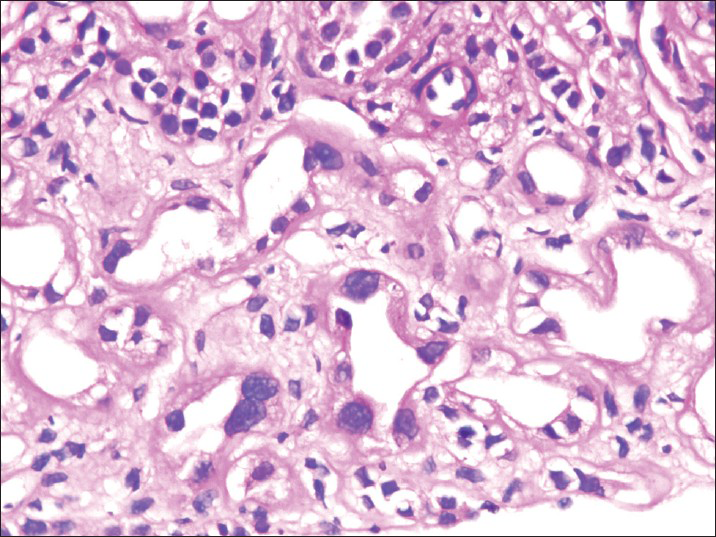
- Karyomegaly in tubular epithelial cells (H and E, ×400)

- (a) Segmental sclerosis and tubular atrophy (Periodic acid Schiff, ×200). (b) Tubular atrophy and interstitial inflammation (H and E, ×200). (c) Interstitial fibrosis (Masson's Trichrome, ×200)
Immunohistochemical stains for BK virus and cytomegalovirus were negative. Ki-67 index was low. Immunofluorescence did not reveal any significant immune deposits.
He is under regular follow-up and his last creatinine has increased to 1.2 mg/dl.
Discussion
KIN is a rare disorder characterized by enlarged tubular epithelial cell nuclei and chronic interstitial nephritis. These patients present with renal impairment and extra renal manifestations are rare. Histologically, presence of interstitial nephritis along with karyomegaly in the tubular epithelial cells is characteristic of this disorder. Karyomegalic cells have been identified in various tissues like astrocytes, schwann cells, intestinal smooth muscle and bile duct epithelium. No clinical significance has been identified with these changes. Sclare[4] described pneumopathy, charaterized by pulmonary karyomegalic cells at autopsy. Transient elevation of liver enzymes is described.[5] Pathogenesis of this disease is unclear and controversial. Toxins or viral infections are suggested as cause of this disorder.[2] Exposure to herbs, ochratoxins is also implicated.
McCulloch et al., reported KIN in three adolescent patients treated with ifosfamide for Ewings sarcoma.[6] All patients progressed to renal failure. A familial clustering is known and frequency of human leukocyte antigen (HLA)-A9 and HLA-B35 haplotypes suggest the possibility of genetic susceptibility.[7] Another genetic defect on chromosome 6, linked to major histocompatibility complex locus is also suspected.[8]
Abnormal deoxyribonucleic acid ploidy, distribution with high ploidy values is described. Exome sequency study by Zhou et al., identified mutations in FAN1 as cause of KIN.[9] The morphological alterations in renal epithelial cells are thought to be the initial damage caused by either chemicals or viral agents, which in susceptible individuals lead to disruption of cells. Advanced glomerulopathy as initial change is unlikely. Immunofluorescence and histological findings are negative in a large series presented by Bhandari et al. In contrast our case clinically presented with nephrotic syndrome and also had histological evidence of focal segmental glomerulosclerosis (FSGS). As there is no positive family history or drug history and exposure to toxins, this case probably represents a sporadic occurrence. KIN is an increasingly recognized entity although is underdiagnosed. It is important to diagnose this entity as it is a progressive disorder leading to irreversible renal damage. The case under discussion has both glomerular and tubulo-interstitial pathology. Common etiological factors for both these lesions are toxins and drugs, patient on repeated questioning denied any history of drug or herbal medicine intake. Heredo-familial occurrence is described both in FSGS and KIN. There is no history of renal disease in the family and it unlikely to be familial because the chromosomal abnormalities described are different for both. Spoendlin et al.,[8] reported four patients who were asymptomatic initially, but later experienced progressive renal failure. This particular case under discussion also initially presented with normal renal function and on follow-up was showing gradual deterioration.
Conclusions
We present a rare case of KIN who initially presented with normal renal parameters; interestingly, this patient's clinical presentation was of nephrotic syndrome whose biopsy also revealed FSGS. This association of KIN and FSGS is not described in the literature. Hence, we think that the patient has idiopathic FSGS with an incidental KIN, which is known to occur sporadically.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Extreme dysplasia in renal epithelium of a young woman dying from hepatocarcinoma. J Pathol. 1974;113:147-50.
- [Google Scholar]
- Systemic karyomegaly associated with chronic interstitial nephritis. A new disease entity? Clin Nephrol. 1979;12:54-62.
- [Google Scholar]
- Karyomegalic tubulointerstitial nephritis: A rare cause of chronic kidney disease. Nephrourol Mon. 2011;3:201-3.
- [Google Scholar]
- A case of systemic karyomegaly associated with interstitial nephritis. Ann Med Interne (Paris). 1998;149:291-4.
- [Google Scholar]
- Karyomegalic-like nephropathy, Ewing's sarcoma and ifosfamide therapy. Pediatr Nephrol. 2011;26:1163-6.
- [Google Scholar]
- Karyomegalic nephropathy: An uncommon cause of progressive renal failure. Nephrol Dial Transplant. 2002;17:1914-20.
- [Google Scholar]
- Karyomegalic interstitial nephritis: Further support for a distinct entity and evidence for a genetic defect. Am J Kidney Dis. 1995;25:242-52.
- [Google Scholar]
- FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. 2012;44:910-5.
- [Google Scholar]




