

Translate this page into:
Plasma exchange in Immunoglobulin A nephropathy with thrombotic microangiopathy and acute cortical necrosis
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
A 25-year-old female presented with decreased urine output, deranged renal function, thrombocytopenia, and hemolytic anemia. Kidney biopsy was consistent with thrombotic microangiopathy with acute cortical necrosis and Immunoglobulin A nephropathy (IgAN). Hemolytic anemia, thrombocytopenia and urine output improved after five sessions of plasma exchange. Renal function showed a delayed recovery and serum creatinine normalized by 3 months. This is first case of successful use of plasma exchange in hemolytic uremic syndrome with cortical necrosis associated with IgAN.
Keywords
Hemolytic uremic syndrome
Immunoglobulin A nephropathy
thrombotic microangiopathy

Introduction
Immunoglobulin A nephropathy (IgAN) is a heterogeneous disease with variable clinical manifestations ranging from asymptomatic hematuria to advanced renal failure. Dialysis-dependent renal failure at presentation in IgAN is usually due to crescentic glomerulonephritis. Thrombotic microangiopathy (TMA) leading to dialysis-dependent renal failure is rare.[1] Although TMA secondary to glomerular diseases has been reported,[1234] TMA with hemolytic uremic syndrome (HUS) and acute cortical necrosis (ACN) in association with IgAN has been rarely reported.
Case Report
A 25-year-old female presented with diffuse dull aching abdominal pain, vomiting and decreased urine output. There was no history of oral ulcers, arthralgia, malar rash, photosensitivity, Raynaud's phenomenon or. Blood pressure was 110/70 mm Hg. On examination, patient appeared pale and had pitting edema in bilateral lower extremities. She was afebrile, and the neurological, cardiovascular, chest and abdominal examination revealed no abnormal findings.
On laboratory evaluation, urine analysis showed 1+ albuminuria with 5–6 red blood cells/high power field. Hematological investigations showed hemoglobin of 9.9 g/dl with 3% schistocytes on peripheral smear examination, total leucocyte count of 9600/mm3 and a platelet count of 3 × 104/mm3. Her coagulation profile showed prothrombin time of 11 s, activated partial thromboplastin time of 22 s and international normalized ratio of 1.0. She had deranged renal function with blood urea nitrogen and serum creatinine of 163 and 6.4 mg/dl respectively. Serum bilirubin was elevated to 2 mg/dl with unconjugated fraction of 1.8 mg/dl. Alanine transaminase, aspartate transaminase and alkaline phosphatase were 67 U/L, 58 U/L and 98 U/L respectively. Her total protein was 6.5 g/dl and serum albumin was 3.6 g/dl. Stool cultures were negative for Escherichia coli and Shigella dysenteriae. Other investigations showed complement factor 3 (C3) and complement factor 4 (C4) of 124 mg/dl (normal range 84–175 mg/dl) and 31.8 mg/dl (normal range 15–50 mg/dl) respectively, lactate dehydrogenase (LDH) was 6209 IU/L (normal range 240–480 IU/L). Hepatitis B surface antigen and Hepatitis C antibody were negative; anti-nuclear antibody by immunofluorescence and antineutrophil cytoplasmic antibody by immunofluorescence and ELISA were negative. Ultrasound showed right kidney and left kidney of 10 cm and 10.5 cm respectively with the loss of corticomedullary differentiation on both sides.
With evidence of hemolytic anemia and renal failure, a diagnosis of HUS was considered and patient received five sessions of plasma exchange. These sessions were done on alternate days and 40 ml/kg of plasma was removed during each session and replaced with fresh frozen plasma with alternate day hemodialysis (HD). After the fifth session of plasma exchange, LDH decreased to 600 IU/L, platelet count improved to 3 × 106/mm3 and urine output improved to more than 1 L/day. However, serum creatinine remained >6 mg/dl and patient required HD. Subsequently percutaneous kidney biopsy was done. Light microscopy revealed evidence of ACN in the form of infarcted glomeruli with severe loss of cellular details of proximal tubular cells and extensive areas of hemorrhage involving 25% of the biopsied area [Figure 1a] and also there was an evidence of TMA in the form of fibrinoid necrosis of one of the arteries with near total occlusion of the lumen [Figure 1b]. Glomeruli from the viable area showed mesangial expansion with increase in cellularity [Figure 2a]. There were no crescents or glomerular basement membrane thickening. Direct immunofluorescence showed the deposition of IgA (3+), C3 (2+), and lambda (3+) in the mesangium and focally along glomerular capillary wall [Figure 2b]. Overall features were suggestive of IgAN with TMA and patchy cortical necrosis. After 2 months, she became dialysis independent, and creatinine reduced to 1.2 mg/dl at 3 months.
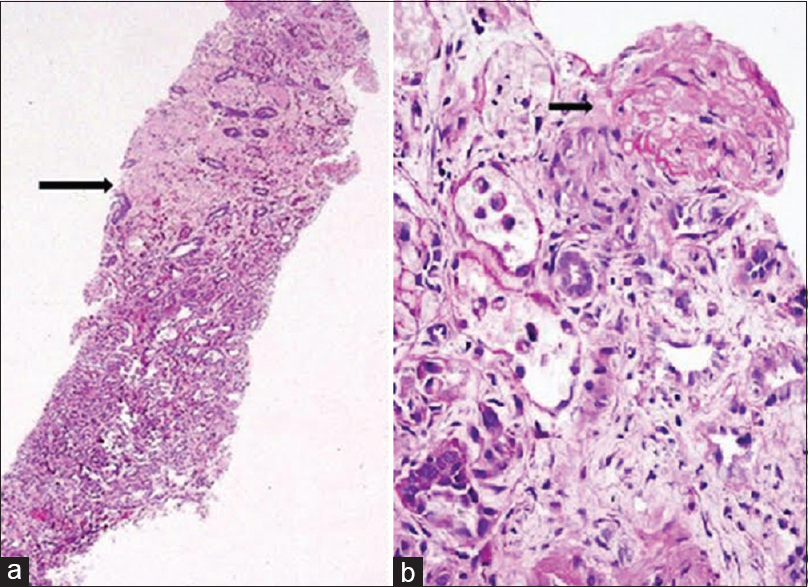
- (a) The core biopsy with half of the cortex showing acute cortical necrosis (arrow) (PAS, ×40). (b) Illustrates the thrombotic microangiopathy in one of the vessels with total occlusion of the lumen (arrow) (PAS, ×400)

- (a) Mesangial expansion and mild mesangial cellularity (H and E, ×400). (b) Direct immunofluorescence reveals granular positivity in the mesangium with immunoglobulin A (3+)
Discussion
Oresentation with renal failure, microangiopathic hemolytic anemia, thrombocytopenia that suggested the diagnosis of HUS. Renal biopsy confirmed TMA with cortical necrosis and underlying mesangial expansion with increased cellularity and IgA deposition. The patient was managed with plasma exchange and had a successful recovery of renal function.
Hemolytic uremic syndrome occurs with different predisposing conditions. In infants and children, diarrheal prodrome precedes HUS (typical HUS), whereas in adolescents and adults, it may present without a prodrome (atypical HUS [aHUS]); both the conditions are thought to be pathogenetically different. Different underlying conditions predispose to aHUS like factor H[4] and I[5] deficiencies, membrane cofactor protein (MCP, now described as CD46)[6] deficiency, pregnancy,[7] malignancy,[8] drugs[9] etc. Endothelial dysfunction is the final common path, which leads to platelet activation, aggregation and arteriolar or capillary occlusion and manifestations of HUS.
Until now, only three cases of HUS in association with IgAN are described. The first reports by Hoshii and Kadowaki[10] and Okabe et al.[11] are published in Japanese language. Morita et al.[1] described a male patient who had proteinuria for 18 years, and presented with hemolytic anemia, thrombocytopenia and renal failure. He was managed with seven sessions of HD and two sessions of plasma exchange. After this treatment, his renal function improved (creatinine came down to 2 mg/dl from 9.6 mg/dl). Subsequently, kidney biopsy was done that was consistent with HUS and IgAN. Renal TMA in patients with IgA nephropathy is a well-defined entity, and many clinicopathological studies have shown it to be a poor marker of renal outcomes.[12] However, the reason why these lesions occur in IgA nephropathy is still unknown and whether intra-renal TMA can initiate HUS is also a debatable issue. A possible explanation could be that primary IgAN is an immune complex-mediated glomerulonephritis and that intra-renal arterial and arteriolar lesions might be related to immune abnormality, including C3 deposition which leads to endothelial cell injury and initiates HUS in genetically susceptible patients (defects in complement regulatory genes), with local intravascular coagulation, and fragmentation of erythrocytes passing through the affected vessels. In our patient, there was a histopathological evidence of TMA involving intra-renal artery with C3 deposits. HUS is also described in association with other chronic glomerular diseases such as focal segmental glomerulosclerosis (FSGS),[2] membranous glomerulonephritis,[3] membrano-proliferative glomerulonephritis (MPGN).[13] Almost all the cases, which are described above, shared one feature, the nephrotic state at the onset of HUS. However, in this present case, there was no evidence of nephrotic syndrome. Manenti et al.[14] published six cases of aHUS with underlying glomerulopathy, one of these patients had FSGS, two MPGN type 1, one C3 glomerulonephritis and two systemic small vessel vasculitis (one granulomatosis with polyangiitis, one Henoch–Schoenlein purpura). Of these six patients, five had the risk haplotypes of complement regulatory genes.[14] In our patient, genomic analysis was not done to look for complement regulator genes due to lack of availability. The data so far showed that the patients had an initial diagnosis of glomerulopathy, which includes IgAN and other glomerulopathies and later showed a shift to aHUS. However, our patient presented with features of both TMA and IgAN simultaneously on kidney biopsy. In our patient, it is difficult to say whether IgAN is responsible for TMA or it is a mere trapping of IgA in the mesangium, which was noticed by chance. However, significant mesangial expansion and cellularity on light microscopy substantiate the possibility of IgAN. It could be probable that the patient had complement dysregulation as demonstrated by C3 on immunofluorescence and had superimposed IgA nephropathy, which facilitated the clinical presentation as HUS. Several investigators used various modalities of treatment in patients with HUS with underlying glomerulopathy (other than IgAN), which includes steroids, rituximab, plasma exchange and ecluzimab without any significant response.[14] As mentioned previously, Morita et al.[1] managed the patient with HUS and IgAN with plasma exchange and showed significant response. Our patient's renal function also normalized with five sessions of plasma exchange. Studies have shown that patients with HUS with TMA involving arteries have a higher likelihood of progressing into ACN compared with patients with predominant glomerular TMA.[1516] As mentioned before, our patient had an evidence of TMA involving intra renal artery.
The evaluation of the case is limited by lack of genetic studies and antibody assay to factor H/I and MCP. To conclude, HUS may be rarely reported secondary to glomerular disease like IgA nephropathy and the disease may respond very well to conventional treatment of HUS that is, plasma exchange.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Hemolytic uremic syndrome associated with immunoglobulin A nephropathy: A case report and review of cases of hemolytic uremic syndrome with glomerular disease. Intern Med. 1999;38:495-9.
- [Google Scholar]
- Recurrent haemolytic uraemic syndrome in a boy with focal and segmental glomerulosclerosis. Eur J Pediatr. 1992;151:791-2.
- [Google Scholar]
- Hemolytic uremic syndrome associated with glomerular disease. Am J Kidney Dis. 1989;13:144-7.
- [Google Scholar]
- Factor H and atypical hemolytic uremic syndrome: Mutations in the C-terminus cause structural changes and defective recognition functions. J Am Soc Nephrol. 2006;17:170-7.
- [Google Scholar]
- Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:2150-5.
- [Google Scholar]
- The association of pregnancy with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Curr Opin Hematol. 2003;10:339-44.
- [Google Scholar]
- Cancer-associated hemolytic-uremic syndrome: Analysis of 85 cases from a national registry. J Clin Oncol. 1989;7:781-9.
- [Google Scholar]
- Drug-associated thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Curr Opin Hematol. 2001;8:286-93.
- [Google Scholar]
- A case of IgA nephropathy progressed to CRF with association of HUS. Jin Touseki. 1990;29:777-80.
- [Google Scholar]
- A case of HUS with IgA deposition of mesangio-capillary type. J Pediatr Pract. 1985;48:269-73.
- [Google Scholar]
- A clinicopathologic study of thrombotic microangiopathy in IgA nephropathy. J Am Soc Nephrol. 2012;23:137-48.
- [Google Scholar]
- Hemolytic uremic syndrome in a girl, mimicking membranoproliferative glomerulonephritis with a worsening course. Rev Clin Esp. 1984;172:289-92.
- [Google Scholar]
- Atypical haemolytic uraemic syndrome with underlying glomerulopathies. A case series and a review of the literature. Nephrol Dial Transplant. 2013;28:2246-59.
- [Google Scholar]
- Decreasing incidence of renal cortical necrosis in patients with acute renal failure in developing countries: A single-centre experience of 22 years from Eastern India. Nephrol Dial Transplant. 2007;22:1213-7.
- [Google Scholar]
- Prognosis and pathological characteristics of five children with non-Shiga toxin-mediated hemolytic uremic syndrome. Pediatr Int. 2007;49:196-201.
- [Google Scholar]




