

Translate this page into:
Collapsing glomerulopathy in a case of anti-neutrophil cytoplasmic antibody associated vasculitis
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Collapsing glomerulopathy (CG) is a pathological entity characterized by collapse and wrinkling of glomerular tuft, podocyte dedifferentiation and hyperplasia. CG may be idiopathic or secondary to other diseases. CG has been described with IgA nephropathy, membranous glomerulopathy, diabetic nephropathy, and lupus nephritis. However, till date there is no report of CG in association with the anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis (AAV). Here, we present a case of CG that developed during follow-up in a case of AAV with biopsy proven pauci-immune glomerulonephritis.
Keywords
Anti-neutrophil cytoplasmic antibody associated glomerulonephritis
collapsing glomerulopathy
pauci-immune glomerulonephritis

Introduction
Collapsing glomerulopathy (CG) is a distinct pathologic entity characterized by segmental or global glomerular capillary collapse, podocyte swelling and hyperplasia, tubulocystic changes, and tubulointerstitial inflammation. Although CG was classically described with human immunodeficiency virus (HIV) infection, it has been increasingly recognized in association with various other conditions that can cause podocyte injury leading to their dedifferentiation and proliferation. Apart from infections, drugs and malignancies, CG has also been described with primary and secondary glomerular diseases including IgA nephropathy, membranous glomerulopathy, diabetic nephropathy, and lupus nephritis. Irrespective of the primary pathology, the presence of glomerular collapse portends a poor prognosis.[1] Till date, no case of CG in association with the anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis (AAV) has been described. We hereby report a case of CG that developed during the follow-up of a patient with AAV and biopsy proven pauci-immune glomerulonephritis.
Case Report
A 41-year-old male presented with 3 months history of intermittent low grade fever, dry cough, pain, and swelling in small joints of hands, feet, and bilateral knees with no early morning stiffness. There was no history of upper respiratory tract symptoms, hemoptysis, chest pain, shortness of breath, pedal edema, lower urinary tract symptoms, hematuria, abdominal pain, vomiting, loose stools, and headache or visual symptoms. In the 3rd month of illness, the patient developed nausea and vomiting and was admitted in local hospital. The evaluation revealed hemoglobin of 6.6 g/dl and serum creatinine of 4 mg/dl. His urine routine and microscopic examination showed 2 + proteinuria, 2–4 pus cells, and 10–12 erythrocytes. Further evaluation revealed positive antinuclear antibody and cytoplasmic ANCA (cANCA) by indirect immunofluorescence (IIF) and positive anti-proteinase 3 (anti-PR3) ANCA by enzyme-linked immune sorbent assay (ELISA). During the course of hospitalization, his serum creatinine increased rapidly to 9 mg/dl, and he was initiated on hemodialysis. He was also given two units of packed red cell transfusion and three intravenous (i.v.) pulses of injection methylprednisolone (1 g each) before referring to our center for further management.
At presentation, he had a pulse rate of 92/min and his blood pressure was 150/90. He also had mild pallor, while other general and systemic examination was normal. He had a drop in hemoglobin from 9 g/dl to 7 g/dl over a period of 3 days; however, there was no associated hemoptysis. A high-resolution contrast tomography of the chest was done which showed patchy areas of dense, ground glass opacities in both lungs with septal thickening suggestive of alveolar hemorrhage. His repeat immunological work-up performed revealed 3+ cANCA positivity by IIF and anti-PR3 ANCA positivity by ELISA while the anti-glomerular basement membrane antibodies were negative. He continued to be oliguric with a serum creatinine of 7 mg/dl and was prescribed regular hemodialysis. Kidney biopsy revealed 12 glomeruli, of which three had cellular crescents and nine fibrocellular crescents along with glomerulitis. The underlying tuft was normal in three glomeruli while it was sclerosed in the rest. Tubules showed patchy acute injury and focal erythrocyte casts. The interstitium showed mild diffuse fibrosis and chronic inflammatory cell infiltration. Blood vessels did not show any diagnostic abnormality. On immunofluorescence, the biopsy was negative for immunoglobulins and complement [Figure 1].
- Photomicrograph showing fibrocellular crescents in the glomeruli with underlying normal tuft (H and E, ×10)
A diagnosis of AAV with pauci-immune crescentric glomerulonephritis and diffuse alveolar hemorrhage was made, and he was prescribed seven sessions of alternate day therapeutic plasma exchange (60 ml/kg), which was replaced with fresh frozen plasma and albumin. He was also given i.v. cyclophosphamide along with oral steroids 1 mg/kg/day. The dose of i.v. cyclophosphamide was according to his estimated glomerular filtration rate (eGFR). He received three doses of i.v. cyclophosphamide at 2 weekly intervals followed by next four doses at 3 weekly intervals. Oral steroids were continued at a dose of 1 mg/kg/day for 8 weeks, followed by gradual tapering to a dose of 5 mg/day at the end of 16 weeks. The patient responded to the treatment with a decline in serum creatinine to 1.8 mg/dl within 1-month of the treatment, which he continued to maintain for next 4 months.
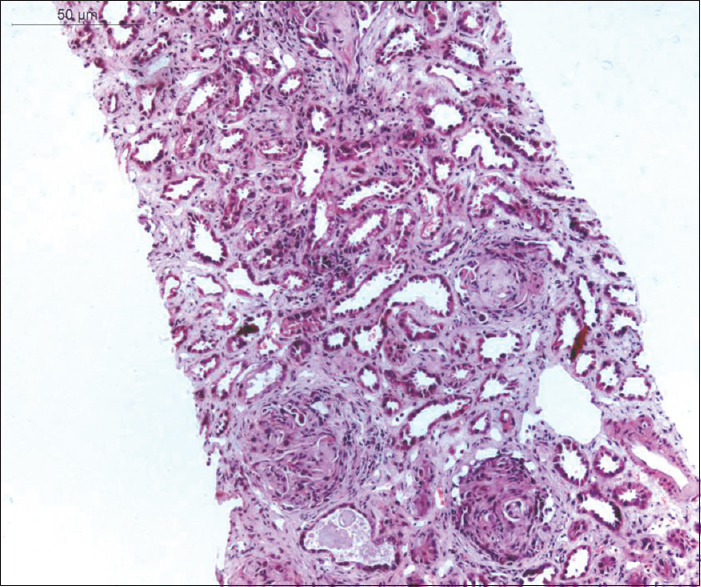
Two weeks after the last dose of cyclophosphamide, he started developing worsening of hypertension along with pedal edema. Investigations revealed an increase in serum creatinine to 3 mg/dl, with a hemoglobin of 9.2 g/dl and serum albumin 4 g/dl. His urine examination revealed 2+ protein and few erythrocyte casts and his 24 h proteinuria was 2.4 g. A relapse of disease was suspected, and he was subjected to repeat renal biopsy to decide about further management. The biopsy showed nine glomeruli, of which one was globally sclerosed; one was normal, and one was incomplete. Five glomeruli showed fibrous crescents and one glomerulus showed the collapse of the glomerular tuft along with podocyte hyperplasia. The tubules showed erythrocyte casts and granulocytic casts with mild patchy tubular atrophy, mild interstitial fibrosis (interstitial fibrosis and tubular atrophy [IFTA] about 15–20%) and chronic inflammation. Some of the arteries and arterioles showed partial to near total vaso-occlusion due to intimal thickening and medial hypertrophy. No evidence of active vasculitis was noted [Figure 2]. The immunofluorescence showed five glomeruli, all of which were negative for immunoglobulins and complement. There were no cytopathogenic changes suggestive of cytomegalovirus (CMV) infection and staining for parvovirus was negative. Serum polymerase chain reaction for CMV deoxyribonucleic acid was also negative. ANCA performed at this time was negative by both IIF and ELISA. As there was no obvious cause of the collapse and considering the chronicity of renal biopsy, he was started on maintenance immunosuppression with azathioprine 75 mg/day along with steroids 5 mg/day. During next 3 months of follow-up, he continues to have 3+ proteinuria and serum creatinine of 4 mg/dl.

- Photomicrograph showing proliferating podocytes with protein absorption droplets and collapsed tuft. Inset shows KI 67 positive podocytes confirming the proliferation of podocytes (periodic acid Schiff ×40, Inset immunohistochemistry KI 67 ×40)
Discussion
The term collapsing glomerulopathy (CG) was first used by Weiss et al. in 1986 to describe the renal pathological lesions in HIV-negative patients with severe proteinuria and rapid progression to renal failure.[2] Later, CG was described as one of the five variants of focal segmental glomerulosclerosis (FSGS) under the Columbia classification and was defined as the presence of segmental capillary tuft collapse (wrinkling and folding) in at least one glomerulus, in association with podocyte hypertrophy and/or hyperplasia.[3] In 2007, Barisoni et al., proposed a taxonomy for the podocytopathies and classified CG separately from FSGS. Being podocytopathies, both CG and FSGS are characterized by podocyte injury, however, CG differs from FSGS in terms of podocyte dedifferentiation and proliferation instead of podocyte depletion. CG lesions are defined by “pseudo-crescent” formation and by the collapse of the capillary loops. While glomerular lesions are the hallmark of CG, the tubulointerstitial disease is also an important component of this entity and is often out of proportion to the extent of glomerular sclerosis.[4] The present case had mild IFTA, while the characteristic protein resorption droplets in the tubules were absent, probably due to a lesser degree of proteinuria.
Clinically, CG has a male predominance, racial predisposition in blacks and presents as massive proteinuria, hypertension, renal dysfunction, and can progress rapidly to dialysis requiring renal failure.[5] However, a significant proportion of patients (15–20%) may present with subnephrotic proteinuria[6] as was seen in our case.
Etiologically, CG can be classified as idiopathic, genetic, and reactive forms.[4] The most commonly encountered among these are the reactive forms, which may be associated with infections, drugs, auto-immune diseases, hematological malignancies, and some other diseases [Table 1]. Among the glomerular diseases, CG has been described with IgA nephropathy,[10] membranous glomerulonephritis (MGN),[11] systemic lupus erythematosus (SLE),[12] and diabetic nephropathy[13] and is associated with worse outcomes.

Karoui et al. described 11 patients with CG coexisting with IgA nephropathy. Clinically, they presented with advanced renal insufficiency and heavy proteinuria (mean 4 g/24 h), with half of them having malignant hypertension. Of the 10 patients with follow-up data, nine finished on hemodialysis while the remaining patient had an eGFR of 31 ml/min/1.73 m2 at 22 months postbiopsy despite aggressive treatment.[14]
In a case series, of three patients with CG coexisting with MGN, one patient progressed to end-stage renal disease (ESRD) within 2 years while another patient had a rapid decline in renal function from stage three to ESRD within 3 months despite treatment.[11]
In a retrospective series, of 19 patients with SLE (16 patients) or SLE-like (3 patients) disease with CG, 95% of patients had the nephrotic syndrome at presentation. Of the 13 patients followed up for 21 months, 54% progressed to ESRD and 38% had some degree of renal dysfunction and proteinuria despite treatment; only 8% achieved normal renal function.[12]
In a retrospective study, of 534 kidney biopsies performed in diabetic patients for either increasing proteinuria or deteriorating renal function, or both, 5% HIV-negative patients were found to have CG superimposed on diabetic nephropathy, of which 92% had nephrotic range proteinuria. Follow-up data were available in 17 patients, out of which 13 (77%) developed ESRD within 7 months of biopsy, while 23% had increasing serum creatinine with stable proteinuria at 5–24 months follow-up.[13]
Anti-neutrophil cytoplasmic antibody AAV with renal involvement usually presents with rapidly progressive glomerulonephritis along with microscopic hematuria, subnephrotic proteinuria, and elevated serum creatinine. Hallmark histologic lesions include crescentic glomerulonephritis and fibrinoid necrosis, while the immunofluorescence is typically negative for all immunoglobulins and complements, giving the terminology of pauci-immune glomerulonephritis. With time, although the necrotic glomerular lesions of pauci-immune crescentic glomerulonephritis heal as segmental or global sclerosis;[15] CG, however, has not been described. Thus, our case is the first case of AAV associated with CG in the absence of any active vasculitis or thrombotic microangiopathy or any evidence of drugs or infections implicated in the pathogenesis of CG. The development of collapse, in this case, may be attributed to the vascular occlusion evident in the biopsy that may be the result of underlying hypertension.
The patients of CG are managed in a variety of ways in the absence of any specific recommendations for its treatment. Variable success rates have been reported with steroids and other cytotoxic agents (cyclophosphamide and cyclosporine), with a dismal complete remission rate of 9.6% and partial remission rate of 15.2%.[5] The risk of progression to ESRD is quite high in CG with the reported incidence being 50–100% in most series.[6] As there is no effective therapy for CG, a number of experimental drugs has been tried with variable success in experimental models. These include retinoic acid derivatives, small molecule inhibitors of cyclin-dependent kinases, and small molecule inhibitors of inflammatory pathways controlled by nuclear factor-kappa B and cyclooxygenase-2.[1]
In the present case, in the absence of any active lesions or vasculitis, we thought that collapse was probably secondary to hypertensive vascular occlusion and decided not to treat aggressively the patient and maintenance immunosuppression was started. However, the malignant nature of the collapsing lesions is evident in the form of persistent proteinuria and gradually rising creatinine level.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Nephrotic syndrome, progressive irreversible renal failure, and glomerular “collapse”: A new clinicopathologic entity? Am J Kidney Dis. 1986;7:20-8.
- [Google Scholar]
- Pathologic classification of focal segmental glomerulosclerosis: A working proposal. Am J Kidney Dis. 2004;43:368-82.
- [Google Scholar]
- A proposed taxonomy for the podocytopathies: A reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol. 2007;2:529-42.
- [Google Scholar]
- Collapsing glomerulopathy: The expanding etiologic spectrumof a shrinking glomerular lesion. 2012. Topics in Renal Biopsy and Pathology. Available from: http://www.intechopen.com
- [Google Scholar]
- Development of focal segmental glomerulosclerosis after anabolic steroid abuse. J Am Soc Nephrol. 2010;21:163-72.
- [Google Scholar]
- Collapsing glomerulopathy associated with natural killer cell leukemia: A case report and review of the literature. Am J Kidney Dis. 2011;58:855-9.
- [Google Scholar]
- Collapsing glomerulopathy in sickle cell disease: a case report. J Med Case Rep. 2011;5:71.
- [Google Scholar]
- Focal segmental glomerulosclerosis in IgA nephropathy: A result of primary podocyte injury? Kidney Int. 2011;79:581-3.
- [Google Scholar]
- Collapsing glomerulopathy coexisting with membranous glomerulonephritis in native kidney biopsies: A report of 3 HIV-negative patients. Am J Kidney Dis. 2003;42:591-5.
- [Google Scholar]
- Collapsing glomerulopathy in 19 patients with systemic lupus erythematosus or lupus-like disease. Clin J Am Soc Nephrol. 2012;7:914-25.
- [Google Scholar]
- Collapsing glomerulopathy superimposed on diabetic nephropathy: Insights into etiology of an under-recognized, severe pattern of glomerular injury. Nephrol Dial Transplant. 2014;29:392-9.
- [Google Scholar]
- Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. II. Light microscopic and clinical studies. Kidney Int. 2011;79:643-54.
- [Google Scholar]
- Heptinstall's Pathology of the Kidney (6th ed). Philadelphia, PA: Lippincott Williams & Wilkins; 2007.




