

Translate this page into:
Recurrent truncating mutations in alanine-glyoxylate aminotransferase gene in two South Indian families with primary hyperoxaluria type 1 causing later onset end-stage kidney disease
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Primary hyperoxaluria type 1 is an autosomal recessive inborn error of metabolism due to liver-specific peroxisomal enzyme alanine-glyoxylate transaminase deficiency. Here, we describe two unrelated patients who were diagnosed to have primary hyperoxaluria. Homozygous c.445_452delGTGCTGCT (p.L151Nfs*14) (Transcript ID: ENST00000307503; human genome assembly GRCh38.p2) (HGMD ID CD073567) mutation was detected in both the patients and the parents were found to be heterozygous carriers. Our patients developed end-stage renal disease at 23 years and 35 years of age. However, in the largest series published from OxalEurope cohort, the median age of end-stage renal disease for null mutations carriers was 9.9 years, which is much earlier than our cases. Our patients had slower progressions as compared to three unrelated patients from North India and Pakistan, who had homozygous c.302T>C (p.L101P) (HGMD ID CM093792) mutation in exon 2. Further, patients need to be studied to find out if c.445_452delGTGCTGCT mutation represents a founder mutation in Southern India.
Keywords
Alanine-glyoxylate transaminase
chronic
India
nephrolithiasis
primary hyperoxaluria type 1
renal insufficiency

Introduction
Primary hyperoxaluria type 1 (PH1) (OMIM 259900) is an autosomal recessive inborn error of metabolism due to deficiency of liver-specific peroxisomal enzyme alanine-glyoxylate transaminase (AGT). Until date, only three genetically confirmed cases have been reported from North India[1] (tests undertaken in The Netherlands) and none from South India. Here, we report two unrelated patients from South India having recurrent mutations in exon 4 of alanine-glyoxylate aminotransferase (AGXT) gene (tests done in our institution).
Case Reports
Case 1
A 20-year-old lady from Tamil Nadu, born of nonconsanguineous parents, was referred for confirmation of suspected primary hyperoxaluria. Two weeks prior, she was admitted elsewhere with advanced uremia and was initiated on twice weekly hemodialysis. She was apparently normal until 16 years of age when she was evaluated for the acute abdomen and found to have recurrent urolithiasis along with nephrocalcinosis. Twenty-four-hour urine oxalate was elevated (69 mg/day, normal range: 4–31 mg/24 h). She received conservative management and remained asymptomatic until the current episode. Birth and developmental history were unremarkable.
At a presentation, she was dehydrated, had a body mass index of 22.5 and had a urine output of 1200 ml/day. On examination, she had horizontal creases on the anterior aspect of incisor teeth. The rest of the examination was within normal limits. X-ray abdomen revealed multiple radiopaque stones almost filling both the kidneys [Figure 1a]. Computed tomography revealed bilateral medullary nephrocalcinosis with focal caliectasis [Figure 1b]. Twenty-four-hour urine oxalate levels were normal for her three siblings. Sanger sequencing of AGXT gene was performed using previously described primer set in ABI 3500 sequencer.[2] The patient was found to be homozygous for c.445_452delGTGCTGCT (p.L151Nfs*14) [Transcript ID ENST00000307503; human genome assembly RCh38.p2) (HGMD ID CD073567)[3] truncating mutation [Figure 2b], and both the parents were heterozygotes [Figure 2c and d] for the same mutation [Figure 2a is showing wild type sequence for comparison]. This is a previously described pathogenic mutation in PH1. This is a previously described pathogenic mutation in PH1. She was planned for combined liver and renal transplantation.
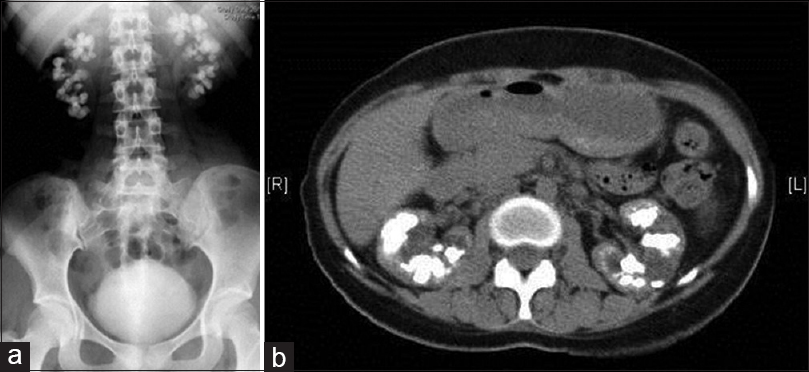
- (a) X-ray of kidney-ureter-bladder region of patient 1 showing both the kidney studded with radiopaque stones; (b) Computed tomography abdomen of patient 1 showing multiple renal stones and nephrocalcinosis

- Sequencing electrophoregram showing (a) wild type sequence, (b) mutant homozygous sequence in the index case and (c,d) heterozygous sequences in the parents of patient 1
Case 2
A 36-year-old man, first-born of nonconsanguineous parentage from Kerala, was referred to Medical Genetics services for genetic confirmation of PH1. He had frequent episodes of abdominal colic due to nephrolithiasis starting from the age of 24 for which he was treated with extracorporeal shock wave lithotripsy of left kidney. Chemical analysis revealed calcium oxalate crystals. He developed hypertension and subsequently became dialysis dependent. There was no family history of similar illness. He was also found to be homozygous for c. 445_452delGTGCTGCT (p.L151Nfs*14) [Transcript ID ENST00000307503; human genome assembly GRCh38.p2) (HGMD ID CD073567)[3] truncating mutation and mother were heterozygous for the same. Father could not be tested because he had passed away earlier.
Discussion
The definitive diagnosis of PH1 is made by estimation of AGT enzyme activity in liver biopsy specimens. This technique is cumbersome as the sample needs to be shipped frozen to the handful of laboratories around the world and is invasive. Molecular diagnostics are proposed to overcome these limitations.[45] The AGXT gene encoding the AGT protein is located at the telomeric end of the long arm of chromosome 2, 2q37.3 and is composed of 11 exons covering approximately 10 kb of genomic DNA.[6] Over 157 pathogenic mutations are listed in Human Genome Mutation Database professional version (accessed on 13.04.2015). Screening for three most common mutations, c.33dupC (p. Lys12GlnfsTer156), c.508G>A (p. Gly170Arg), and c.731T>C (p. Ile244Thr), enabled a molecular diagnosis in 34.5% cases of Northern European descent.[5] There are case reports of newer mutations associated with PH1 from various parts of the world.[78] The overlapping eight nucleotide deletion (GCTGCTGT) to the p.L151Nfs*14 mutation in exon 4 (GTGCTGCT) described here has been previously reported in six unrelated Asians from the United Kingdom.[9] Our patients had end-stage renal disease (ESRD) at 23 years and 35 years of age. However, the median age of ESRD for null mutations carriers was 9.9 years in the largest series published from OxalEurope cohort[10] which is much earlier than our cases. On the other hand, in the previously described three unrelated patients from North India and Pakistan carrying c.302T>C (p.L101P) (HGMD ID CM093792) homozygous mutation in exon 2 of AGXT gene had an onset below 7 years of age[1] which roughly corresponds with OxalEurope cohort.[10]
Therefore, the present case represents the first report of molecularly proven PH1 from Southern India. The mutation exemplifies rare instance of truncating mutation with residual biological activity. We need to genotype more patients from South India to find out if p.L151Nfs*14 is a founder mutation in South India/whether exon 4 represents a deletion hotspot for these patients. The delay in age of onset in our patient points to other factors such as diet and other compensatory metabolic pathways in glyoxylate metabolism.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Common mutation underlying primary hyperoxaluria type1 in three Indian children. Indian J Nephrol. 2012;22:459-61.
- [Google Scholar]
- Comprehensive mutation screening in 55 probands with type 1 primary hyperoxaluria shows feasibility of a gene-based diagnosis. J Am Soc Nephrol. 2007;18:1905-14.
- [Google Scholar]
- Primary hyperoxaluria type 1: Update and additional mutation analysis of the AGXT gene. Hum Mutat. 2009;30:910-7.
- [Google Scholar]
- Genetic analysis – A diagnostic tool for primary hyperoxaluria type I. Pediatr Nephrol. 2002;17:896-8.
- [Google Scholar]
- Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias. Kidney Int. 2004;66:959-63.
- [Google Scholar]
- Characterization and chromosomal mapping of a genomic clone encoding human alanine: Glycosylate aminotransferase. Genomics. 1991;10:34-42.
- [Google Scholar]
- Mutation spectrum of primary hyperoxaluria type 1 in Tunisia: Implication for diagnosis in North Africa. Gene. 2013;527:316-20.
- [Google Scholar]
- Mutational analysis of AGXT in two Chinese families with primary hyperoxaluria type 1. BMC Nephrol. 2014;15:92.
- [Google Scholar]
- Selected exonic sequencing of the AGXT gene provides a genetic diagnosis in 50% of patients with primary hyperoxaluria type 1. Clin Chem. 2007;53:1216-21.
- [Google Scholar]
- Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int. 2014;86:1197.
- [Google Scholar]




