

Translate this page into:
Unusual cause of glomerular deposition disease: Collagenofibrotic glomerulopathy
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Collagenofibrotic glomerulopathy is a rare condition characterized by deposition of Type III collagen fibers in the subendothelial space and mesangium of the glomerulus. Only 17 cases have been reported from India. A definite diagnosis can only be established when typical histological findings are supported by electron microscopy. It is characterized by indolent progression and has no definitive therapy.
Keywords
Collagenofibrotic glomerulopathy
glomerular deposition disease
nephrotic syndrome

Introduction
Collagenofibrotic glomerulopathy (CG) is a rare condition characterized by deposition of Type III collagen fibers in the subendothelial space and mesangium of the glomerulus.[12] It was first reported in 1979 by Arakawa et al.[3] It was initially considered as a variant of Nail–Patella syndrome in view of similar histological findings but without skeletal abnormalities.[45] The patients range in age from 2 to 66 years, with no sex predilection.[6] The most common clinical presentation is proteinuria with or without associated nephrotic syndrome, with minor alterations in renal function.[7] Only 17 cases have been reported from India.[6] We report a case of this rare entity.
Case Report
A 63-year-old man, hypertensive for ten years, nondiabetic, a vegan, and with no significant past or family history, presented with anasarca since 2½ months. On physical examination, his blood pressure was 160/100 mm Hg and fundus showed grade two hypertensive retinopathy. Laboratory investigations revealed macrocytic anemia (hemoglobin 10.7 g/dl), 24 h urine protein of 3.9 g, and serum albumin of 2.7 g/dl. The serum creatinine (0.8 mg/dl), lipid profile, complement levels, thyroid profile were within normal range. There was an associated Vitamin D deficiency (9.6 ng/ml) and Vitamin B12 deficiency (135 mcg/dl). Myeloma screening was negative. Ultrasound of abdomen showed normal sized kidneys with increased echotexture. Viral screen for HIV, hepatitis B virus, and hepatitis C virus was negative, and coagulation profile was normal.
The renal biopsy [Figure 1] showed 15 glomeruli, two of whom were sclerosed. Viable glomeruli were enlarged in size with deposition of pale eosinophilic material in the mesangium forming nodules. This material was negative for Periodic Acid Schiff (PAS), Congo red and positive with silver methenamine. Masson trichrome showed increased collagen within the nodules. The glomerular basement membrane (GBM) was mildly thickened and showed focal reduplication. There was no evidence of tubule-interstitial chronicity or inflammation. Direct immunofluorescence of renal biopsy with IgM, IgA, IgG, C3, C1q, and kappa and lambda light chains were negative ruling out the presence of immune complex deposits. These features were suggestive of a possible CG, which was further confirmed with electron microscopy. Electron microscopy [Figure 2] showed markedly enlarged glomerulus with lobular arrangement and diffuse foot process effacement. The mesangium and capillary lumina revealed subendothelial deposits of large fibers (97 nm width) which are curvilinear and with disorganized arrangement and periodicity suggestive of collagen fibers; confirming the diagnosis of collagenous glomerulopathy.
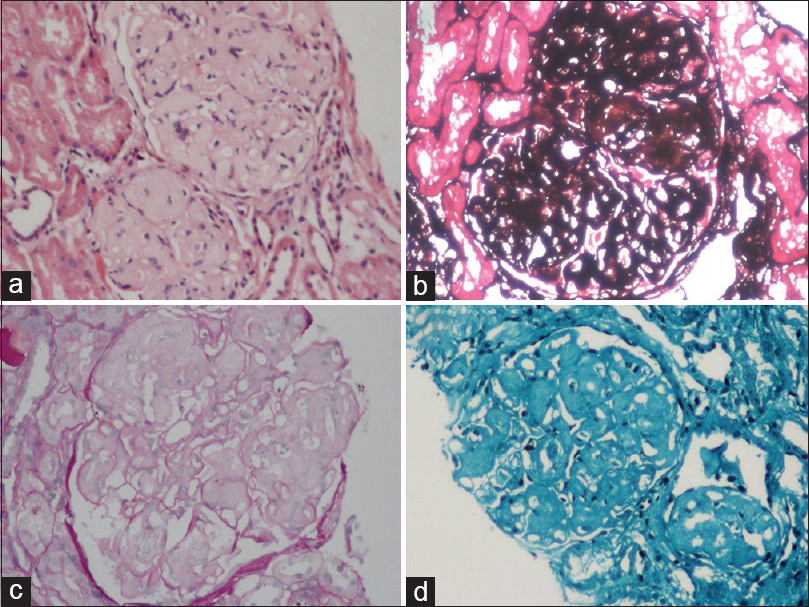
- (a) The glomerular enlargement with mesangial expansion with the formation of nodules. The nodules were (b) positive for silver stains (c) negative with Periodic Acid Schiff stain and (d) collagen deposition with Masson trichrome stain

- The electron microscopic study of renal biopsy specimen fixed using glutaraldehyde shows the presence of subendothelial deposits of large fibers which are curvilinear and with disorganized arrangement suggestive of collagen fibers; confirming the diagnosis of collagenous glomerulopathy
No electron dense deposits were identified. The lamina densa of GBM was unremarkable. All these features confirmed the diagnosis of CG.
Discussion
CG is recognized as a distinct entity and is relatively common in Asian countries.[6] Ethnic/genetic factors play an important role in etiopathogenesis. There are two fundamental theories regarding the genesis of this disease. First is that normal human glomeruli lack collagen III, and it is postulated that mesangial cells are the endogenous source of collagen III. The second theory is that this disease is a systemic disorder with abnormal metabolism of collagen III.[8] The incidence of CG is highest between the fourth and seventh decades, notably among Asian patients; while those reported in Europe are children.[9] The most common clinical presentation is edema and/or persistent proteinuria that may be a nephrotic range (about 60% of patients). Our patient is a 63-year-old who presented with generalized edema and had nephrotic range proteinuria. Hypertension is seen in the two-third of cases at the time of presentation. Our subject had hypertension since 10 years and may not be a manifestation of CG. Anemia may be noticed even before the development of renal dysfunction. Occasionally, microangiopathic hemolytic anemia has been documented in children. Our patient had macrocytic anemia secondary to Vitamin B12 deficiency as he was a strict vegan. Only 17 cases of CG have been reported from India to the best of our knowledge. Table 1 summarizes the clinical profile of published cases of CG in literature. Extrarenal symptoms are absent, unlike in cases of Nail–Patella syndrome. The natural history of the disease is variable, but it is progressive in most patients. Children are more likely to progress to end-stage renal failure. Successful renal transplantation without a recurrence has been documented in one case.[7] The only specific test for CG is an estimation of procollagen III N-terminal peptide levels. Procollagen peptide levels or immunohistochemistry for collagen typing was not performed in our patient. Renal function tests are usually normal or slightly increased at presentation. Urine examination occasionally shows microscopic hematuria and proteinuria but no active urine sediment. Autoimmune workup is usually negative, and monoclonal immunoglobulins are absent in serum/urine.

Light microscopy[6] shows lobular bland appearing glomeruli due to the global expansion of the mesangium with thickening of the peripheral capillary walls. The mesangial expansion is due to the accumulation of amorphous, weakly PAS- and silver-positive material. However, Congo red and thioflavin stains are completely negative. The thickened capillary walls show focal reduplication; however, PAS and methenamine silver stains clearly highlight that the capillary basement membranes are normal, and thickening of the wall is due to subendothelial deposition of pale amorphous material. In the advanced stage, capillary lumens are narrowed by the expanded mesangium and thickened capillary walls and glomeruli show a nodular appearance suggestive of diabetic nephropathy or monoclonal immunoglobulin deposition disease. However, unlike these two entities, the nodular lesions are weakly PAS positive or PAS negative in CG. Immunofluorescence staining is usually negative. Immunohistochemistry for specific collagen types shows abundant staining for Type III collagen.
Electron microscopy is essential for differential diagnosis of immunofluorescence negative deposition diseases such as CG, fibronectin glomerulopathy, and diabetic glomerulosclerosis. But in standard electron microscopy, the lesion is not readily visible; staining with tannic acid, phosphotungstic acids make the fibers clearly visible. These fibers are curved, frayed (unlike the interstitial fibers which are straight). They have a transverse band with a periodicity of 43–65 nm, characteristically associated with collagen III. These fibers are absent in lamina densa of the basement membrane, which is typical of Nail–Patella syndrome. Various degrees of foot process effacement is usually seen.
No specific treatment has been described. Supportive measures include control of hypertension and diuretics to relieve edema. Renal replacement therapy may be required for patients with the end-stage renal disease. Although very few patients have received a transplant, to date, none have shown recurrence of the disease.[7] The role of systemic glucocorticoid treatment in CG is doubtful although it may be useful in dermatological manifestations.
To conclude, collagneofibrotic glomerulopathy is an important and rare cause of nodular glomerulosclerosis. The special stains and electron microscopy are essential for exact characterization of this glomerulopathy. This report highlights the details of such a case.13
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Primary glomerular fibrosis: A new nephropathy caused by diffuse intra-glomerular increase in atypical type III collagen fibers. Clin Nephrol. 1990;33:155-9.
- [Google Scholar]
- Collagen type III glomerulopathy: A new idiopathic glomerular disease. Am J Nephrol. 1991;11:422-9.
- [Google Scholar]
- Idiopathic mesangio-degenaerative glomerulopathy (Japanese) Jpn J Nephrol. 1979;21:914-5.
- [Google Scholar]
- The nail-patella syndrome-pathogenesis of the kidney lesion. Birth Defects Orig Artic Ser. 1974;10:57-9.
- [Google Scholar]
- Nail patella-like renal lesions in the absence of skeletal abnormalities. Am J Kidney Dis. 1982;1:237-40.
- [Google Scholar]
- Collagenofibrotic glomerulopathy – Case report with review of literature. Indian J Nephrol. 2011;21:52-5.
- [Google Scholar]
- Collagenofibrotic glomerulopathy: Clinicopathologic overview of a rare glomerular disease. Am J Kidney Dis. 2007;49:499-506.
- [Google Scholar]
- Glomerular diseases with organized deposits. In: Jennette JC, Silva FG, Olsan JL, D’Agati VD, eds. Hiptenstall's Pathology of Kidney (7th ed). Philadelphia: Wolters Kluwer; 2015. p. :1029-31.
- [Google Scholar]
- Collagen type III glomerulopathy: A new type of hereditary nephropathy. Pediatr Nephrol. 1993;7:354-60.
- [Google Scholar]
- Banded collagen in the kidney with special reference to collagenofibrotic glomerulopathy. Ultrastruct Pathol. 2010;34:68-72.
- [Google Scholar]
- Collagenofibrotic glomerulopathy in association with Hodgkin's lymphoma. Saudi J Kidney Dis Transpl. 2011;22:126-9.
- [Google Scholar]
- Collagenofibrotic glomerulopathy – Case report with review of literature. Indian J Nephrol. 2011;21:52-5.
- [Google Scholar]




