

Translate this page into:
Renal Tubular Acidosis, Osteopetrosis, and Cerebral Calcification: A Rare Syndrome Caused by Carbonic Anhydrase II Deficiency
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Sir,
Carbonic anhydrase deficiency II is a rare disorder, which presents classically as a triad of osteopetrosis, renal tubular acidosis (RTA), and cerebral calcification.
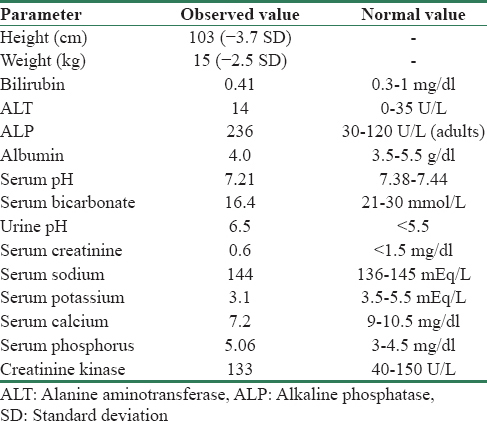
A 7-year, 10-month-old boy, product of a consanguineous marriage, presented with short stature and a recent fracture of left forearm following a trivial trauma. There was no significant family history. His height was 103 cm (−3.7 standard deviation) [Table 1]. He had dysmorphic facies such as prominent forehead, thick lower lip, and blue sclera. He had 24 teeth; his temporary teeth had not shed yet. There were no features of rickets or tetany. On investigation, he had normal complete blood count and liver and kidney function tests. Investigations also revealed hypocalcemia, systemic acidosis, and alkaline urine (systemic pH of 7.21 and urine pH of 6.5 in the presence of normal renal functions) [Table 1]. Radiological survey showed hyperdense bones in forearm and lower limbs with a fracture in the left forearm [Figure 1]. Non-contrast computed tomography head showed bilateral basal ganglia calcification. Thus, in view of classical triad of osteopetrosis, RTA, and cerebral calcification, a diagnosis of carbonic anhydrase II deficiency was considered. Sanjad-Sakati syndrome, Kenny-Caffey syndrome, and Kirk-Richardson syndrome may present with a similar phenotype, these usually are not associated with RTA. Carbonic anhydrase II deficiency is a rare disease with varied presentation and most commonly presents as a triad of RTA, osteopetrosis, and cerebral calcification.[1] Other manifestations include short stature, mental retardation, anemia with ovalocytosis, retinal changes, and multiple cranial nerve palsies.[2] These diverse manifestations are due to omnipresent location of the enzyme, and the degree of involvement depends on the extent of enzyme expression in a particular organ. RTA is usually a mixed type with features of both proximal and distal types.[3] Carbonic anhydrase II has important role in both proximal and distal tubules in kidney for generation of H+ and HCO3−; thus, features of both proximal and distal RTA are found. Osteopetrosis results from decreased activity of osteoclasts.[4] Cerebral calcifications usually develop after 5 years of age and are more common in basal ganglia region and sometimes in periventricular subcortical white matter. Exact mechanism of the development of cerebral calcifications is not clear, but carbonic anhydrase II has a definite role in cerebrospinal fluid secretion.[2]

- (a) X-ray left forearm showing increased density, with a classic chalk stick fracture of left ulna. (b) x-ray knees also suggestive of diffuse increase in bone density. (c) Noncontrast computed tomography head showing bilateral basal ganglia calcification
Other features of this disorder include typical facies such as craniofacial disproportion with a prominent forehead, small mouth, micrognathia, and thick lower lip. Optic atrophy and retinal atrophy have also been found in some patients. Hematological disorders including anemia, thrombocytopenia, and leukopenia are typically prominent in severe variety of recessive disease. Other biochemical abnormalities include raised brain isoform of creatine kinase and acid phosphatase. These patients may also have hypocalcemia with secondary hyperparathyroidism.[5] Our patient had short stature, RTA, cerebral calcifications, osteopetrosis, dysmorphic facies, bilateral maculopathy, raised creatine kinase, and hypocalcemia, thus making the diagnosis of carbonic anhydrase II deficiency very likely.
The identification of structural gene mutation underlying carbonic anhydrase II deficiency provides a simple and accurate molecular technique for diagnosis of this disease. The gene is located on chromosome 8 (8q22). There are seven exons in this gene, and mutations have been reported in each of them. It has also been found that mutations vary as per geographical area also – Middle Eastern people have different mutations compared to their European, American or Japanese counterparts.[3]
Carbonic anhydrase deficiency is a very rare disease with wide range of clinical manifestations. Diagnosis is very likely if RTA is associated with osteopetrosis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Osteopetrosis, renal tubular acidosis and basal ganglia calcification in three sisters. Am J Med. 1980;69:64-74.
- [Google Scholar]
- Variable clinical presentation of carbonic anhydrase deficiency: Evidence for heterogeneity? Eur J Pediatr. 1990;149:337-40.
- [Google Scholar]
- The carbonic anhydrase II deficiency syndrome: Osteopetrosis with renal tubular acidosis and cerebral calcification. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Diseases (7th ed). New York: McGraw-Hill; 1995. p. :4113-24.
- [Google Scholar]
- Morphological evidence of reduced bone resorption in osteopetrotic (op) mice. Am J Anat. 1982;163:157-67.
- [Google Scholar]




