

Translate this page into:
Systemic Lupus Erythematosus with Linear IgA Bullous Dermatosis and Renal Vascular Lesions: An Extremely Rare Association
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
We report a rare case of systemic lupus erythematosus presenting initially with cutaneous manifestations of linear IgA bullous dermatosis. Later the patient developed renal abnormalities due to thrombotic microangiopathy and lupus nephritis with inflammatory necrotizing vasculitis. Paucity of immune deposits was observed on Immunofluorescence. This association of SLE with these cutaneous and renal lesions is rarely reported in the literature.
Keywords
Linear IgA bullous dermatosis
lupus
thrombotic microangiopathy
vasculitis

Introduction
Systemic lupus erythematosus (SLE) is known for the gamut of cutaneous and renal manifestations. The cutaneous lesions in SLE are heterogeneous, which include bullous and non-bullous lesions. The reports on coexisting SLE and linear IgA bullous dermatosis (LABD) are sparse. Both being autoimmune disorders, their exact association is not clear. Furthermore, despite the high frequency of renal involvement, renal vascular involvement is very rare, the most common being uncomplicated vascular immune deposits. Here, we report a case of young female presented with LABD and renal involvement initially in the form of thrombotic microangiopathy (TMA) and later as lupus nephritis with inflammatory necrotizing vasculitis.
Case Report
A 20-year-old female presented with malar rash, discoid rash, photosensitivity, and arthritis. On investigation, she had leukopenia and tested positive for ANA and ds-DNA. Having satisfied 7 out of the 11 ACR diagnostic criteria, she was diagnosed as a case of SLE. Urine examination was unremarkable and serum creatinine was 0.5 mg/dl. She was started on steroids (1 mg/kg body weight) and hydroxychloroquine (400 mg/day) but was not responsive. Hence, methotrexate (15 mg weekly) was started. Two years later, she presented with a generalized rash for 1 week. There was a history of intake of Non-Steroidal anti-inflammatory drugs 3 days before the onset of rash. On examination, erythematous lesions with overlying fluid-filled blisters were noted. There was no hypertension. Skin biopsy was done and sent for light microscopy and immunofluorescence (IF). The skin biopsy showed a subepidermal blister containing neutrophils [Figure 1a]. Homogeneous linear IgA deposition along the basement membrane zone of the normal skin was observed on direct IF [Figure 1b]. Thus, a diagnosis of LABD was made.
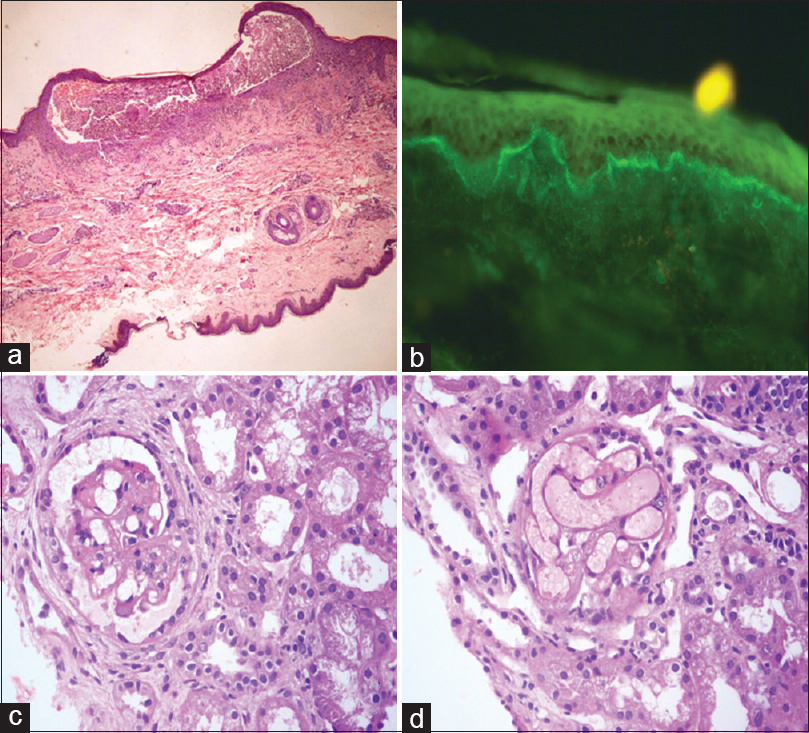
- (a) Subepidermal blister containing neutrophils (H and E, ×100). (b) Linear IgA deposits in basement membrane zone on direct immunofluorescence (×400). (c) Glomerulus with mesangiolysis (H and E, ×400). (d) Congested glomerular tuft (H and E, ×400)
Other laboratory investigations revealed hemoglobin (Hb) of 8.4 g/dl, 2+ albumin in the urine along with plenty of red blood cells (RBCs), 24-h urine protein of 750 mg/24 h, and serum creatinine of 0.67 mg/dl. C3 and C4 were both decreased with levels of 63.2 mg/dl and 10 mg/dl, respectively. Renal biopsy done at this stage revealed occasional bloodless glomeruli with mesangiolysis [Figure 1c] and some with dilated, congested capillaries [Figure 1d]. The histopathological features were suggestive of TMA. There were no proliferative/necrotizing/chronic lesions. The tubules, interstitium, and blood vessels were unremarkable. IF was negative. Antiphospholipid antibody was negative. The patient was started on mycophenolate mofetil (1 g/day).
After 8 months, the patient developed bilateral lower limb swelling, joint pain, and breathlessness. Urine examination showed 3+ albumin with many RBCs and a few white blood cells. Serum creatinine was 3.92 mg/dl, blood urea nitrogen of 131 mg/dl, Hb of 5.4 g/dl, and platelet count of 80,000/cmm. Both direct Coombs test and indirect Coombs test were negative, and C3 and C4 levels were low. Echo showed a pericardial effusion while her chest X-ray showed left lower zone consolidation. After 2 days of admission, she became oligo-anuric and her blood pressure was 180/100 mmHg while her serum creatinine rose to 5 mg/dl. She was initiated on hemodialysis, started on cyclophosphamide (500 mg every 2 weeks), and pulsed with methylprednisolone (0.5 g/day for 3 days). At this point, the clinical diagnosis was Class III/IV lupus nephritis with crescentic transformation because of doubling serum creatinine. A second renal biopsy was done at this stage. Light microscopy revealed global endocapillary proliferation and mild neutrophilic infiltration [Figure 2b]. Cellular and fibrocellular crescents were present (33%) [Figure 2c]. The arteries and arterioles with fibrinoid necrosis, transmural neutrophilic infiltrate, and karyorrhexis were evident [Figure 2a]. Chronicity features were present. Diffuse, global, granular deposits of IgG, IgA, IgM, C3, and C1q of 1+ intensity were observed in the mesangium and capillary loops on direct IF [Figure 2d-f]. The final histopathological diagnosis was lupus nephritis Class IV (A/C) with inflammatory necrotizing vasculitis with an activity index of 7/24, chronicity index of 8/12.
To summarize, the patient presented with a congregation of the LABD, TMA and Class IV lupus nephritis with inflammatory necrotizing vasculitis.
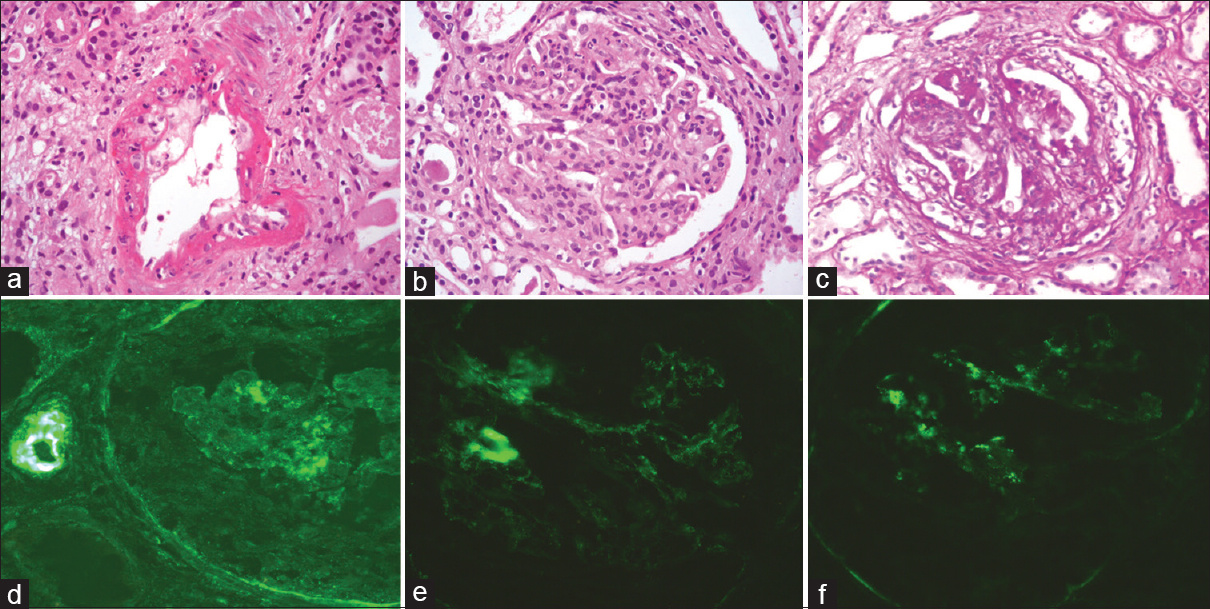
- (a) Inflammatory necrotizing vasculitis of interlobular artery (H and E, ×400). (b) Global endocapillary proliferation (H and E, ×400). (c) Cellular crescent (H and E, ×400). (d-f) Direct immunofluresence with IgG, C3, and C1q immunostains, respectively, showing weak positivity in the mesangium and capillary loops (×400)
Discussion
SLE is known to present in myriad forms. In SLE, cutaneous manifestations are more common and are extremely heterogeneous. LABD is characterized by subepidermal bulla with neutrophils and linear IgA deposits in the basement membrane. LABD in SLE is considered as a nonspecific bullous lesion that may occur very rarely. Although the etiology is largely unknown, LABD had been reported in association with drugs, infections, and connective tissue disorders. Lee et al.[1] and Tobón et al.[2] have reported similar cases. However, in contrast to this report, both the cases were diagnosed as SLE after the initial presentation with cutaneous lesions. Both LABD and SLE being autoimmune disorders the possibility of a common immunopathogenic mechanism between these two needs to be explored.
Lupus nephritis, defined as a quintessential immune complex-mediated disease, can present with a gamut of histological changes involving various renal compartments. The vascular lesions in lupus nephritis have different morphological manifestations with the least common being lupus vasculitis. Although the vascular lesions are not included in the International Society of Nephrology/Renal Pathology Society classification, it tends to carry a poor prognosis. The vascular lesions are generally overlooked. Renal TMA is rare in lupus and can be seen in any class of lupus nephritis. Charney et al.[3] studied five cases of lupus nephritis with TMA. They concluded that proliferative and necrotizing glomerulonephritis may occur in the presence of TMA. In our case, the initial IF was negative. Causes of TMA in SLE include antiphospholipid antibodies (APLA), scleroderma, and malignant hypertension. This patient tested negative for APLA, and there was no evidence of hypertension/systemic thrombosis at the time of biopsy.
Vasculitis is much rarer in lupus nephritis accounting for 0.3%–2.8% of cases.[45] It is histologically indistinguishable from antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis. In the present case, one might argue that this might be an ANCA-associated pauci-immune glomerulonephritis with vasculitis. However, the presence of endocapillary proliferation and orderly crescents are diagnostic of immune complex-mediated disease. Furthermore, ANCA-associated glomerulonephritis is characterized by extensive segmental fibrinoid necrosis which though present was not the most striking feature. In this case, IF revealed a full-house pattern but with very much less intensity than what is usually seen in a case of lupus nephritis. Li et al.[6] and Akhtar et al.[7] had reported similar lupus nephritis cases with absent immune deposits in electron microscopy. There was no vasculitis and was negative for ANCA. Finally, another plausible explanation for the paucity of immune deposits could be prior treatment with drugs such as cyclophosphamide and methylprednisolone. However, in this case, a combined etiology of both lupus nephritis and ANCA-associated glomerulonephritis cannot be ruled out. ANCA testing was not performed as the patient was initiated on plasmapheresis. Sen and Isenberg[8] discovered that around 20% of the patients with SLE show seropositivity with ANCA and chiefly perinuclear-ANCA on indirect IF. On ELISA, the rates of seropositivity were much lower and commonly observed antigens were lactoferrin, cathepsin G and myeloperoxidase.
As lupus vasculitis has an poor prognosis, early and aggressive course of immunosuppressive therapy is essential; hence, these lesions should be promptly reported.
Conclusion
This rare assemblage of cutaneous and renal vascular lesions poses a dilemma for pathologist and clinician. Reporting of subsequent similar cases is imperative to explore the common pathogenic mechanisms and to address the issues of a standardized treatment regimen.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- A case of linear IgA bullous dermatosis associated with systemic lupus erythematosus. Ann Dermatol. 2016;28:660-2.
- [Google Scholar]
- Linear IgA bullous dermatosis associated with systemic lupus erythematosus: A case report. Clin Rheumatol. 2008;27:391-3.
- [Google Scholar]
- “Pauci-immune” proliferative and necrotizing glomerulonephritis with thrombotic microangiopathy in patients with systemic lupus erythematosus and lupus-like syndrome. Am J Kidney Dis. 2000;35:1193-206.
- [Google Scholar]
- Renal vascular lesions as a marker of poor prognosis in patients with lupus nephritis. Gruppo Italiano per lo Studio Della Nefrite Lupica (GISNEL) Am J Kidney Dis. 1991;18:240-8.
- [Google Scholar]
- Pauci-immune necrotizing lupus nephritis: Report of two cases. Am J Kidney Dis. 1994;23:320-5.
- [Google Scholar]
- Antineutrophil cytoplasmic autoantibodies in systemic lupus erythematosus. Lupus. 2003;12:651-8.
- [Google Scholar]




