

Translate this page into:
Crystalline Nephropathy in Renal Transplant: A Series of 4 Cases
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Crystals are particles of endogenous inorganic or organic composition that can trigger kidney injury when deposited or formed inside the kidney. The most common forms of crystalline nephropathies (CNs) are nephrocalcinosis and oxalate nephropathy. The causes of early allograft dysfunction are changing constantly, and recently calcium oxalate (CaOx) crystal deposition has been added to this list. CaOx deposition in renal allograft is important and probably under-recognized cause of delayed graft function that requires adequate awareness with early intervention to improve the allograft outcome. Here, we describe four cases of irreversible renal graft injury due to CNs.
Keywords
Crystalline nephropathy
hyperoxaluria
renal allograft dysfunction

Introduction
Crystalline nephropathies (CNs) refer to renal parenchymal deposition of crystals leading to kidney damage. The most common forms of CN encountered in renal pathology are nephrocalcinosis and oxalate nephropathy. Less frequent types include urate nephropathy, cystinosis, dihydroxyadeninuria, and drug-induced CN (e.g., caused by indinavir or triamterene). Monoclonal proteins can also deposit in the kidney as crystals and cause tissue damage.[1] Oxalate is the ionic form of oxalic acid and is derived from various animal and plant sources. Oxalate is excreted mainly through the kidneys. Hyperoxaluria is a state of disordered metabolism characterized by an increased urinary excretion of oxalate. The normal daily oxalate excretion in healthy individuals ranges between 10 and 40 mg per 24 h. Concentrations exceeding 40–45 mg per 24 h are considered as clinical hyperoxaluria.[2] This may result from increased endogenous production of oxalate in primary hyperoxaluria (PH), from increased in dietary and intestinal absorption (enteric hyperoxaluria), increased intake of oxalate precursors or alteration in intestinal microflora in secondary hyperoxaluria (SH).[2] The causes of early allograft dysfunction are changing constantly, and recently calcium oxalate (CaOx) crystal deposition has been added to this list.[3] Deposition of CaOx crystal in renal tubules can be seen in >50% of allograft biopsies performed <3 months post-transplant.[4] Although the presence of CaOx crystal in allografts can be benign, when present in moderate intensity, it contributes to increased incidence of acute tubular necrosis and poor allograft survival.[34] Systemic oxalosis should be prevented, but the diagnosis is often delayed in more than 40% of patients. In a survey by Hoppe and Langman, 30% of the patients were diagnosed only when they had already reached end-stage kidney disease (ESKD).[5] In some cases, the diagnosis is made after the disease recurs following renal transplant. Hyperoxaluria continues to be a challenging disease, and appropriate treatment requires a high index of suspicion and timely diagnosis. The aim of this presentation is to underline the causes of crystal nephropathy in allografts kidney and the related pathophysiologic mechanisms, which are involved, along with the description of four cases of irreversible renal graft injury due to CNs.
Case Reports
Case 1
A 33-year-old female was on maintenance hemodialysis (HD) for ESKD of unknown etiology. She had first presented 10 years earlier with renal failure when she was found to have bilateral small kidneys on ultrasound examination. Serum and urinary oxalate were not tested because no obvious indication existed then. She received a renal allograft from a living related donor (mother) with a human leukocyte antigen (HLA) mismatch of two. The patient and the donor did not have personal or family history of urolithiasis and their abdominal radiographs were normal. Post-transplant immunosuppressive (IS) treatment included anti-thymocyte globulin (ATG) antibodies as induction agent and corticosteroids, mycophenolate mofetil (MMF), and tacrolimus as maintenance therapy. After surgery, diuresis occurred immediately and her serum creatinine (SCr) levels fell to 300 μmol/L by the 3rd day. However, on day 6 post transplantation, SCr levels went up to 587 μmol/L with reduced urine output. The blood tacrolimus level at this stage was 12 ng/mL. Doppler ultrasound of the graft was normal and there were no features of ureteric obstruction or vascular thrombosis. She underwent an allograft biopsy, which revealed wide-spread tubular degenerative changes typical of acute tubular necrosis and intratubular oxalate crystals. When viewed under polarized light, the tubular oxalate deposits appeared birefringent [Figure 1]. After the biopsy, the patient admitted to have taken at least 2 g/day of Vitamin C daily for several years. There was no specific indication for Vitamin C, but it was prescribed systematically for all patients in her dialysis unit, probably to optimize iron therapy. A 24-h urinary oxalate measurement performed in the donor was 117 μmol/L (normal values: 100–700 μmol/24 h). Bone marrow aspiration was performed, which showed extensive CaOx deposits. After reviewing the biopsy reports, besides advising to avoid high-oxalate foods, we prescribed diuretics (furosemide 1000 mg/day to augment urine output hoping to decrease supersaturation of CaOx and crystal formation) and pyridoxine 300 mg/d to try and eliminate the excess oxalate load. She also received daily HD sessions; however, there was no improvement in her graft function, and the patient returned to maintenance dialysis.
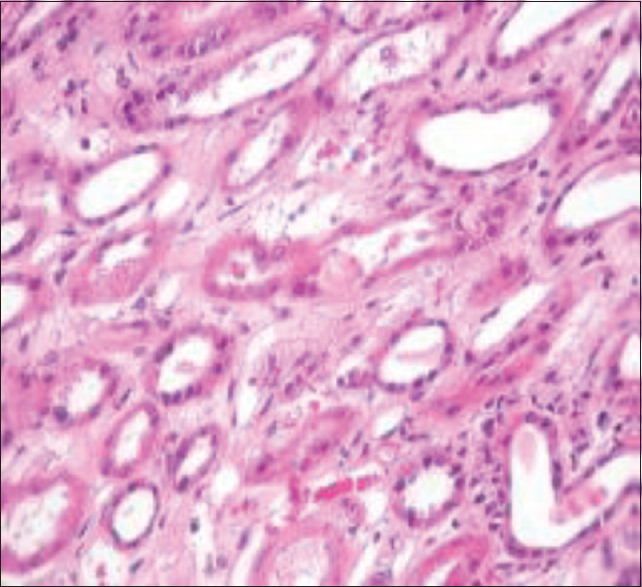
- Proximal tubules with diffuse degenerative changes typical of acute necrosis and tubular calcium oxalate crystals (H and E, ×200)
Case 2
A 50-year-old man was diagnosed as ESKD due to unknown etiology since 2007. He had a strong family history of urolithiasis in his father and sister. However, they were not evaluated. He received a preemptive living-related kidney transplant in 2010. The donor was his mother with 3 HLA mismatches. She did not have a history of urolithiasis, and her abdominal radiograph was normal. Posttransplant immunosuppression treatment included induction with anti-thymoglobulin antibodies, and he was maintained on corticosteroids, MMF, and cyclosporin. Posttransplant, the patient developed delayed graft function (DGF) and slow recovery of allograft function with reduction of SCr to 400 μmol/L over 9 days. The cyclosporine trough level was 150 ng/ml. Doppler ultrasound of the graft was normal, and there were no features of obstruction or vascular thrombosis. He underwent an allograft biopsy, which revealed medullary tissues without tubulitis. Twenty-eight days after transplantation, evolution was marked by spontaneous decrease in SCr to 155 μmol/L. However, the patient developed four episodes of septicemia (Escherichia coli) due to a vesicoureteral reflux Stage IV requiring ureteric reimplantation. He was treated with imipenem and ciprofloxacin. At 18 months posttransplant, the patient was hospitalized due to increasing SCr at 250 μmol/L. Ultrasound examination of the graft and venous duplex of renal vessels were normal. The second allograft biopsy showed tubular atrophy with the presence of CaOx crystals deposition (multicolored birefringence polygonal crystals under polarized light) [Figure 2]. Re-examination of the first graft biopsy confirmed the absence of birefringent deposits in polarized light. In the donor, 24-h urine examination for oxaluria was negative. In view of CaOx deposition, clinical evaluation performed for secondary hyperoxaluria (enteric hyperoxalosis, MMF-induced diarrhea, excessive oral intake of oxalate-containing foods including Vitamin C, ethylene glycol intoxication, or use of methoxyflurane), which was found to be negative. When SCr was 400 μmol/L, urinary pH was 7.2.

- Tubular calcium oxalate deposits appear birefringent under polarized light (H and E, ×200)
Case 3
The patient was a 28-year-old man treated with maintenance hemodialysis since 1998 because of ESKD of an unknown etiology discovered in 1997. Renal biopsy was not performed as both kidneys were small at presentation. Neither self nor family history of urolithiasis or renal disease was reported. In 1999, the patient received a living-related kidney transplant. The donor was his 51-year-old mother, who had a good health with no medical history or physical abnormality at the pretransplantation check-up. Immunosuppression treatment composed of ATG for 10 days, cyclosporin, corticosteroids, and azathioprine. Because of persistent anuria after surgery, duplex ultrasound examination was performed and showed no blood flow in the graft vein. Surgical exploration 6 h after graft found a pleated graft vein and it was corrected. Graft biopsies were performed at the same time. It showed a normal histological appearance with no features of rejection. Twenty-five days after transplantation, urine output improved despite persistent graft dysfunction with a SCr of 700 μmol/l. A second allograft biopsy, performed at day 33, failed to show any features consistent with acute or chronic rejection. Subsequently, graft magnetic resonance imaging examination suggested partial cortical necrosis. However, graft function improved (SCr decreased to 500 μmol/L) and he was free of dialysis. Five months later, the patient was hospitalized because of increment of SCr to 950 μmol/L, while urine flow was maintained at about 1.5 l/24 h. Ultrasound examination of the graft and duplex ultrasound examination of renal vessels were normal. Cyclosporin monitoring was at the therapeutic range. Acute rejection was suspected and treatment with 3 methyl-prednisolone pulses was instituted empirically without a satisfactory response. A third allograft biopsy was performed and showed mild chronic tubule-interstitial changes with CaOx crystal deposits within the interstitium and lumena of the renal tubules. Re-examination of the first graft biopsy identified few CaOx crystal deposits. IS treatment was withdrawn and HD was restarted. Blood and urine oxalic acid in the patient and donor were slightly increased. Serum oxalic acid was 0.073 mmol/L and 0.068 mmol/L, respectively (normal <0.01 mmol/L). In the donor's urine, oxaluria was detected at a level of 0.77 mmol/L (normal <0.4 mmol/L).
Bone marrow examination failed to show oxalate deposits in either the patient or his mother. Native kidney, graft kidney, and mother's kidney showed no nephrocalcinosis or urolithiasis on radiological evaluation. Neither genetic studies nor liver enzyme activity measures were performed.
Case 4
A 34-year-old man was diagnosed with ESKD secondary to chronic interstitial nephropathy. History did not reveal personal or family history of urolithiasis or parental consanguinity. He received a renal allograft from a living-related donor (father) with a three HLA mismatches. Immunosuppression was induced with ATG, and he was maintained on corticosteroids, MMF, and cyclosporin. Despite having good diuresis postoperatively, serum creatinine remained at 350 μmol/L. Duplex ultrasound examination was normal. Since SCr further rose to 573 μmol/L by post-transplant day 21, an allograft biopsy was performed. It revealed the presence of CaOx crystal deposits within the renal tubules [Figure 3]. Biochemical analysis of blade biopsy revealed the presence of CaOx monohydrate crystals. Hemodialysis was restarted.

- Calcium oxalate crystal deposition in the renal tubules under polarized light microscopy
Discussion
Oxalate is a metabolic end product that is excreted essentially unchanged in the urine after absorption in the gastrointestinal tract.[6] Hyperoxaluria is a rare metabolic disorder characterized by CaOx deposition in different tissues. The kidney primarily excretes the excess oxalate. Increased urinary excretion of oxalate results in urinary CaOx supersaturation leading to crystal formation, urolithiasis, and/or nephrocalcinosis. CaOx crystals are typically deposited within the renal interstitium and renal tubular cells. When glomerular filtration rate falls below 30–40 ml/min per 1.73 m2, plasma oxalate levels increase because of reduced urinary oxalate excretion.[7] Urinary oxalate is largely derived from the endogenous metabolism of glycine, glycolate, hydroxyproline, and Vitamin C. Hyperoxalosis from any cause converges on a common final pathophysiological pathway of supersaturation of the renal tubular fluid leading to the precipitation of oxalate crystals in the renal interstitium creating an interstitial nephritis, macrophage recruitment and surge in inflammatory mediators ultimately leading to tubular atrophy and acute kidney injury (AKI).[6]
Hyperoxaluria is divided into two categories: primary (PH) and secondary (SH). PH includes a group of rare autosomal recessive disorders. Its estimated prevalence is <3 in one million and an incidence of 0.15 × 106/year.[8] Disease is characterized by overproduction of oxalate. There are three existing forms of the disease (Type I, II, and III) in which the underlying defects have been specifically identified. Type I PH is caused by the mutation of the liver-specific peroxisomal enzyme alanine-glyoxylate aminotransferase (AGT), whereas type II is caused by the mutation of glyoxylate reductase-hydroxypyruvate reductase. Type III PH results from defects in the liver-specific mitochondrial enzyme 4-hydroxy-2oxo-glutarate aldose. The mechanism that causes an increase in oxalate levels remains unclear. In this frame, potentially other forms of PH could be expected to be reported in the future.[8]
Metabolic history and prevalence of secondary oxalosis are less well studied than that of PH.[9] SH is usually the result of conditions, which are characterized by increased intestinal oxalate absorption, including a high-oxalate diet or fat malabsorption (enteric hyperoxaluria) or alterations of the intestinal oxalate degrading microorganisms and genetic variations of the intestinal oxalate transporter.[7] It may also be secondary to vitamin deficiency (thiamine and pyridoxine) or decreased renal excretion of oxalate.[9] Crystal nephropathy may occur also during the administration of medications excreted by the kidney for which urine solubility depends on urine pH. Several drugs, notably acyclovir, methotrexate, indinavir, and sulfonamide are associated with crystalluria and crystal nephropathy. Sulfadiazine and methotrexate tend to precipitate in tubule when urine pH is low.[10]
The differential diagnosis between PH and SH represents the cornerstone for the treatment. SH should be ruled out before investigating for PH. Urinary oxalate excretion rates, combined with clinical findings, are the first steps for diagnosis. Urine testing for oxalate (normal <0.5), glycolate, and glycerate (L-glyceric acid) can help to distinguish PH from SH. In PH, the oxalate urinary excretion rate is much more (>1 mmol/1.73 m2 BSA/day), whereas in the SH, a lower increase is noted (<1.0 mmol/1.73 m2 BSA/day).[7] In patients with renal insufficiency, urinary oxalate will be falsely low. Oxalate will retain in the systemic circulation and plasma oxalate measurements could be helpful in this situation.[7]
The CaOx crystal appears as a colorless crystal predominantly present in the distal tubules and shows bright white polarization characteristics. The presence of oxalate crystals can be overlooked as these are colorless and are visible only on the hematoxylin and eosin stained slides. In addition, they are dissolved away and not readily visible with the other special stains routinely used for kidney biopsy such as periodic acid–Schiff, trichrome, and silver stains. Polarization microscopy does make them visible in a spectacular fashion and readily demonstrates the true extent.[6]
These 4 cases illustrate the importance of recognizing the presence of oxalate crystals as a distinct etiology of renal allograft dysfunction. In case 1, our patient had no personal or family history of lithiasis, and the donor urinary oxalate was normal. These findings are not consistent with a diagnosis of PH. It is very likely that Vitamin C was involved in the development of oxalosis in this patient because she was taking supranormal doses of this drug for more than 10 years. Vitamin C has been increasingly prescribed for a number of indications ranging from the common cold to anemia management in dialysis patients. Clinicians often underestimate the role of Vitamin C in hyperoxalosis. Vitamin C, being a water-soluble vitamin, offers a false sense of safety and although in the vast majority of cases, daily Vitamin C supplementation may not be of concern. However, it should be given with caution in patients with established risk factors for kidney disease. In addition, patients may receive Vitamin C as part of total parenteral nutrition. Previously reported cases series of liver transplant patients suggest that even normal doses of Vitamin C supplementation (≤500–1000 mg/day) may contribute to oxalosis with the presence of additional risk factors such as a diarrheal illness.[6] A dose not exceeding 100 mg after each HD session (300 mg/week) is safe in these patients.[9]
The cause for AKI in the second case could be attributed to CNs secondary to ciprofloxacin. About 30%–60% of the active drug (ciprofloxacin) is excreted in the urine during 24 h. The drug will be eliminated by glomerular filtration and active tubular secretion. The presence of crystal deposits was first reported in experimental studies in animals known to have alkaline urine. Clinical studies have demonstrated that crystalluria after ciprofloxacin treatment are uncommon and occur in alkaline urine (pH > 7.3).[10] In our patient, urine was alkaline (pH = 7.2). As urine acidification may potentially constitute a specific treatment. As urine acidification may potentially constitute a specific treatment, despite in literature crystal nephropathy might occur with acidic urine,[10] medical practitioners should be aware of this rare complication of a commonly prescribed antibiotic.[11]
The two other cases illustrate a fortuitous discovery of interstitial and tubular oxalate deposits in the renal allografts with no history of urolithiasis in either the donor or the recipient. They may be classified as type 3 PH since they did not experience any CaOx calculi. Enzymatic activities of AGT and GR/HPR should have been measured on liver biopsy specimen and genetic analyses have to be performed to support this hypothesis. It is known that CaOx recurs immediately after transplantation.[12] However, this fact cannot explain CaOx deposits in the third case because of the slight increase of oxalemia. In fact, in PH, the increase of serum oxalate is very important because of the association of an over-production with reduction of urinary excretion as a result of poor renal function. Probably, a moderate AGT defect in the mother and her son with the DGF that explain CaOx deposits seen in our patients, apparently these were enhanced following the surgical complications. In the literature, several cases of delayed diagnosis of PH has been reported, even after renal graft failure.[13] We believe that history of renal calculi with chronic interstitial nephropathy justifies the search for oxalosis before kidney transplantation; however, it is not justifiable to investigate each patient with unknown nephropathy for oxalosis.
Conclusions
CaOx deposition in renal allograft is probably under-recognized cause of DGF that requires adequate awareness with early intervention to improve the allograft outcome. The differential diagnosis between PH and SH is the cornerstone for the management of such patients. Early identification and treatment of SH is important to prevent renal allograft dysfunction or graft loss.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Primary and secondary hyperoxaluria: Understanding the enigma. World J Nephrol. 2015;4:235-44.
- [Google Scholar]
- Primary nonfunction of renal allograft secondary to acute oxalate nephropathy. Case Rep Transplant 2011 2011:876906.
- [Google Scholar]
- Early presence of calcium oxalate deposition in kidney graft biopsies is associated with poor long-term graft survival. Am J Transplant. 2005;5:323-9.
- [Google Scholar]
- A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. 2003;18:986-91.
- [Google Scholar]
- Secondary oxalosis induced acute kidney injury in allograft kidneys. Clin Kidney J. 2013;6:84-6.
- [Google Scholar]
- Secondary hyperoxaluria: A risk factor for kidney stone formation and renal failure in native kidneys and renal grafts. Transplant Rev (Orlando). 2014;28:182-7.
- [Google Scholar]
- Secondary oxalosis due to excess Vitamin C intake: A cause of graft loss in a renal transplant recipient. Saudi J Kidney Dis Transpl. 2014;25:113-6.
- [Google Scholar]
- Crystalluria and ciprofloxacin, influence of urinary pH and hydration. Chemotherapy. 1986;32:408-17.
- [Google Scholar]
- Early recurrence of oxalate deposition after renal transplantation in a patient with primary hyperoxaluria type I. Transplant Proc. 1999;31:3219-20.
- [Google Scholar]
- Renal graft failure due to type 1 primary hyperoxaluria. Neth J Med. 2002;60:407-10.
- [Google Scholar]




