

Translate this page into:
Schimke Immuno-osseous Dysplasia: A Case Report
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Schimke immuno-osseous dysplasia (SIOD) is a rare inherited disease characterized by steroid resistant nephrotic syndrome, spondyloepiphyseal dysplasia, and T-cell immunodeficiency. Focal segmental glomerulosclerosis (FSGS) is the most frequent renal pathological finding associated with proteinuria in SIOD. In this case report, we describe a 4.5-year-old boy who presented with nephrotic syndrome and ventricular septal defect followed by tremor in the limbs after-cerebral infarction. It is emphasized that SIOD should be considered in children with wide range of presentation, from growth retardation, steroid resistant nephrotic syndrome, and bone, cardiac, and neurological abnormalities in the late childhood or even adolescence.
Keywords
Cardiovascular abnormalities
kidney diseases
Schimke immuno osseous dysplasia
SMARCAL1
tremor

Introduction
Schimke immuno-osseous dysplasia (SIOD) is a rare disease caused by a biallelic mutation in the SMARCAL1 gene. It is estimated that the incidence of SIOD is 1 in every 1,000,000 to 3,000,000 live births in the United States.[1] Typical clinical and laboratory findings in SIOD include steroid resistant nephrotic syndrome (SRNS), spondyloepiphyseal dysplasia, T-cell immunodeficiency, progressive renal failure, and hypothyroidism. Transient ischemic attacks, cerebral infarction, atherosclerosis, and migraine-like headache are common neurologic manifestations of SIOD.[234] In this article, we describe a 4.5-year-old boy who presented with nephrotic syndrome and other extra renal manifestations from Iran.
Case History
The proband was the second child of healthy consanguineous (3rd degree) parents. Both parents and siblings were healthy and had no developmental or growth disorders. The first pregnancy of his mother was a boy delivered by caesarean section because of oligohydramnios at gestational age of 31 weeks. He died 2 weeks after delivery because of a cardiac anomaly. The cause of death was probably valvular agenesis, but due to the dissatisfaction of parents, no further evaluation was done to understand the exact cause of his death.
The proband was the product of premature delivery with the gestational age of 32 weeks through caesarean section because of intrauterine growth restriction and oligohydramnios (birth weight = 1670 gram, body length = 42 cm, and head circumferences = 32 cm). He was admitted 4 days in neonatal intensive care unit (NICU) because of prematurity and jaundice. In NICU, because of low O2 saturation and abnormal cardiac exam, echocardiography was done, and ventricular septal defect (VSD) and pulmonary hypertension were diagnosed. When he was 4 months old, serum insulin-like growth factor 1, growth hormone, and IGF binding protein level were below the normal range (40 ng/ml, 8.5 U/L, and 1068 ng/ml, respectively). Therefore, growth hormone therapy was started for him, but no response was observed according to his weight and length for age curves. He underwent VSD repairing surgery at the age of 4.5 months. At the age of 2 years, the brain MRI was done for better evaluation of the cause of his growth failure. It showed hypoplastic pituitary gland. There was no sign of brain infarction at this time. When he was 2.5 years old, in routine work up, proteinuria (3+ protein in a random urine sample and 1383 mg protein in the 24-h urine) and hypercholesterolemia (323 mg/dl) were detected, and prednisolone with atorvastatin were started for him with impression of nephrotic syndrome. Kidney biopsy was not done owing to lack of consent from his parents.
His parents discontinued prednisolone at the age of 4 years. After 6 months, because of severe edema and exacerbation of nephrotic syndrome, he received rituximab in another center. After 4 days, he developed tremor in the upper and lower limbs (more significant in the right hand), fever, and mouth aphthous lesion and was admitted in our center.
On physical exam, he had a triangular face, broad nasal bridge, low-set ears, frontal bossing, short trunk, and kyphosis (height = 76 cm, weight = 8.8 kg, and head circumference = 49 cm). In addition, syndactyly of the second and third toes of the left foot, Café au lait spot over the trunk and right undescended testis were detected [Figure 1]. Except for an increase in the cortical echogenicity of both kidneys, abdominal ultrasound did not reveal any other abnormalities. The dental age was delayed compared to chronological age. The bone survey showed delayed bone age, J-shaped sella, generalized osteopenia, shallow acetabulum on both sides without narrow pelvis, bilateral small femoral heads, and flattening of the cervical and thoracic vertebrae.
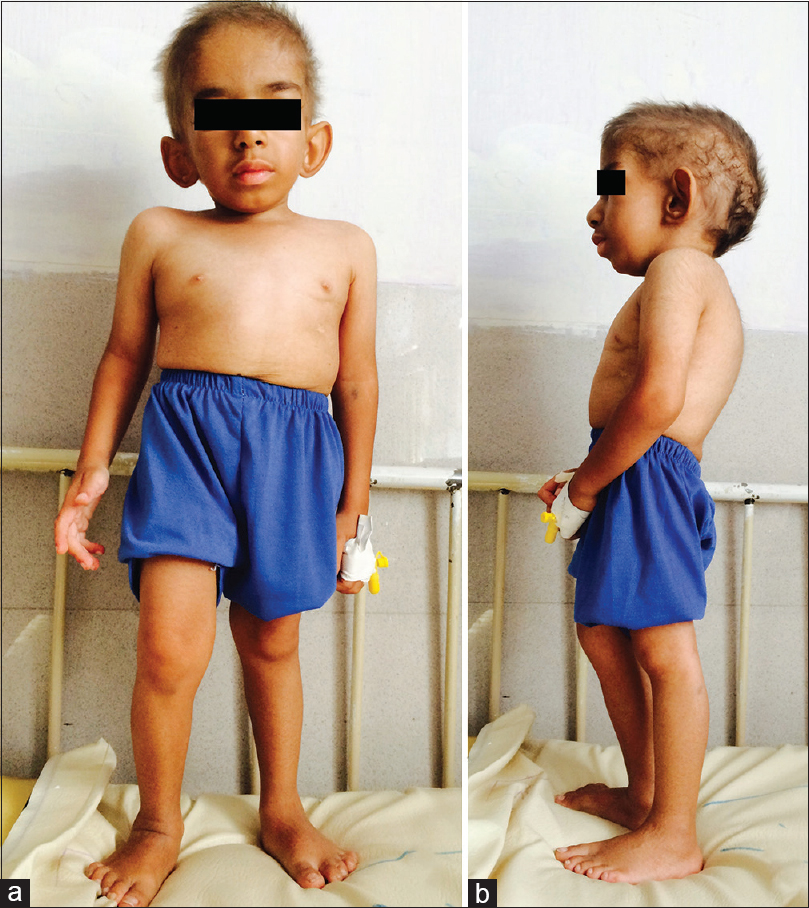
- Anterior (a) and lateral (b) views of the patients show triangular face, broad nasal bridge, low-set ears, frontal bossing, short trunk, and kyphosis
ECG showed 1st degree AV block. Echocardiography showed trivial mitral regurgitation, mild tricuspid regurgitation, an ejection fraction of 77%, and medium to large perimembranous VSD (7.7 mm) associated with bidirectional shunt. Table 1 shows the laboratory findings in this admission. Urine culture showed growth of Escherichia coli. Further workups were not suggestive of endocarditis.
| Laboratory tests | Measured values | Normal range |
|---|---|---|
| Hemoglobin (g/dl) | 10.9 | 12-14 |
| White blood cells (103/μl) | 1.400 | 4000-10000 |
| Polymorph (%) | 80 | |
| Lymphocyte (%) | 10 | |
| Monocyte (%) | 10 | |
| Platelet (103/μl) | 342000 | 150000-450000 |
| Reticulocyte (%) | 1.8 | 0.5-1.5 |
| Erythrocyte sedimentation rate (ESR) (mm/h) | 95 | 0-20 |
| C-reactive protein (CRP) (mg/l) | 5.5 | Under 6 |
| Blood urea nitrogen (BUN) (mg/dl) | 13 | 6-21 |
| Creatinine (mg/dl) | 1 | 0.6-1.2 |
| Alanine aminotransferase (unit/l) | 10 | 7-40 |
| Aspartate aminotransferase (unit/l) | 29 | 12-45 |
| Alkaline phosphatase (unit/l) | 123 | 180-420 |
| Total bilirubin (mg/dl) | 0.5 | 0.2-1.2 |
| Direct bilirubin (mg/dl) | 0.1 | Under 0.4 |
| Albumin (g/dl) | 2 | 3.5-5.2 |
| Sodium (meq/l) | 136 | 136-145 |
| Potassium (meq/l) | 3.9 | 3.5-5.5 |
| Calcium (mg/dl) | 8.1 | 8.8-10.2 |
| Phosphorus (mg/dl) | 3.8 | 2.7-4.5 |
| Triglyceride (mg/dl) | 641 | Under 150 |
| Cholesterol (mg/dl) | 293 | Under 200 |
| Thyroid stimulating hormone (TSH) (mIu/ml) | 2.16 | 0.35-4.94 |
| T4 (µg/dl) | 4.9 | 4.78-11.72 |
| Free T4 (ng/dl) | 1.48 | 0.7-1.48 |
| T3 (ng/dl) | 56 | 58-159 |
| Parathyroid hormone (pg/ml) | 20.1 | 15-68.3 |
| Immunoglobulin G (g/l) | 4.136 | 6.85-18.37 |
| Immunoglobulin M (g/l) | 0.479 | 0.4-1.8 |
| Immunoglobulin A (g/l) | 1.095 | 0.4-1.8 |
| Dihydrorhodamine | 119 | Above 50 |
| Total T-cell (CD3+) (%) | 27 | 43-76 |
| T helper cell (CD4+) (%) | 19 | 23-48 |
| T cytotoxic cell (CD8+) (%) | 8 | 14-33 |
| Natural killer cell (CD16+) (%) | 73 | 4-23 |
| B cell (CD19+) (%) | 0 | 14-44 |
| Gated leukocytes (CD45+) | 79 | |
| (CD4+/CD8+) | 2.38 | 0.9-2.9 |
| Natural killer cell (CD56+) (%) | 73 | 4-23 |
| Monocyte (CD14+) (%) | 18 | 3-6 |
| CD20+ (%) | 0 | 14-44 |
| Adhesion molecules (CD11b) | Normal | |
| ANA (unit/ml) | 3 | Under 10 |
| C-ANCA (unit/ml) | 0.5 | Under 12 |
| P-ANCA (unit/ml) | 0.3 | Under 12 |
| Anti-dsDNA (unit/ml) | 0.3 | Under 16 |
| Urine analysis | Specific gravity: 1.010Protein: 3+ Blood: 3+ Glucose: 1+ RBC: 2-4 WBC: 1-2 Bacteria: few |
|
| 24-h urine protein (mg) | 1237 | Under 140 |
Brain MRI revealed diffuse brain atrophy, periventricular banding and capping as well as pulvinar and bithalamic infarctions extending to the midbrain and infarction in the right inferior cerebellum. In addition, a lacunar infarction in the left side of the deep frontal white matter was seen, which was suggestive of arterial infarction. The brain magnetic resonance angiography (MRA) showed early beaded appearance and irregularity in the anterior cerebral artery and middle cerebral artery, which can be in favor of small vessel disease and mimics early stage of moyamoya [Figure 2].

- Brain MRI revealed diffused brain atrophy and periventricular banding and multiple infarctions in the brain. The MRA showed early beaded appearance and irregularity in the anterior cerebral artery and middle cerebral artery
Because of the characteristic findings, a clinical impression of SIOD was considered, and analysis of SMARCAL1 gene for detection of mutation was performed. The result of the test showed a homozygous, non-synonymous mutation c.[1439C>T] that caused a missense mutation (p.P480L) in the SMARCAL1 protein. The mutation c.[1439C>T] was found by direct sequencing of both sense and antisense strands of the 16 coding and two non-coding exons of SMARCAL1. His parents were heterozygous carriers of the same mutation [Figure 3].

- Analysis of SMARCAL1 gene revealed a homozygous, non-synonymous mutation c.[1439C>T]. (a) Proband, (b) Father, (c) Mother
His parents got discharged him from the hospital with their personal consent, and he died 1 month later. Informed consent form was obtained from parents for publishing the images and clinical data before participation in the study in accordance with the protocol approved by the ethics committee of Shiraz University of Medical Sciences.
Discussion and Conclusions
We present a 4.5-year-old boy with steroid resistant nephrotic syndrome; failure to thrive; and skeletal, cardiac, and neurological anomalies in favor of SIOD. The exact etiology of SIOD is unclear. Mutations of SMARCAL1 gene were detected in about 50% to 60% of patients with SIOD.[1]
FSGS is the most frequent renal pathological finding associated with proteinuria in SIOD. FSGS in individuals with SIOD is usually identified between 1 and 14 years of age and leads to end-stage renal disease in short run.[5] Although FSGS is the leading cause of SRNS, minimal change disease, membranous nephropathy, and mesangial proliferative glomerulonephritis have also been described as the causes of SRNS. In these individuals, there is evidence of temporary reductions in proteinuria by using angiotensin-converting enzyme inhibitors, or cyclosporine-A.[1] Our patient was treated by steroid and rituximab as a case of idiopathic SRNS because the diagnosis of SIOD was not made at that time and after admission in our ward by impression of SIOD, steroid was tapered and he only received supportive therapy.
Functional and anatomical cardiac problems are very uncommon in SIOD.[3] Up to now, VSD was reported only in a 16-year-old girl.[6] Fatty infiltration of the cardiac right ventricular wall was seen in another patient.[3] A prior study revealed the role of SMARCAL1 gene expression in the development of murine cardiomyocytes in the embryonic period.[7]
As a result of recent advances in transplantation and dialysis, life expectancy in these children has increased. Subsequently, cerebrovascular disease has become an important cause of morbidity and mortality.[89] Neurologic presentations in SIOD result from several reasons. Hypertension and hyperlipidemia secondary to renal disease combined with immune dysfunction could predispose the patient to atherosclerosis.[10] Movement disorders such as limbs tremor are uncommon after stroke.[11] Tremor mostly develops after the infarction of the thalamus, especially the posterior nucleus of the thalamus.[12] Therefore, the presence of tremor, as a novel presentation of SIOD, following stroke can be explained.
In conclusion, we reported a 4.5-year-old boy, a case of SIOD, who presented with nephrotic syndrome and suffered from cardiac problem and cerebral infarction. We emphasize that SIOD has a wide range of presentations, from SRNS to cardiac and neurologic abnormalities.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
This article was supported by the Vice Chancellor for Research of Shiraz University of Medical Sciences (grant number: 94-01-18-10110).
Conflicts of interest
There are no conflicts of interest.
Acknowledgment and funding source
The authors wish to thank Dr. Karmela Kamali and Dr. Sepideh Sefidbakht for assistance with MRI and X-ray review, Dr. Majid Fardaei for assistance with DNA extraction, and Dr. Marie Morimoto for performing gene analysis. The authors thank Prof. Nasrin Shokrpour at Center for Development of Clinical Research of Nemazee Hospital for editorial assistance.
References
- Anovel SMARCAL1 mutation associated with a mild phenotype of Schimke immuno-osseous dysplasia (SIOD) BMC Nephrol. 2014;15:41.
- [Google Scholar]
- Schimke immuno-osseous dysplasia: A clinicopathological correlation. J Med Genet. 2007;44:122-30.
- [Google Scholar]
- Eltrombopag (thrombopoietin-receptor agonist) and plasmapheresis as rescue therapy of acute post-renal transplant immune thrombocytopenia in a child with Schimke immuno-osseous dysplasia-case report. Pediatr Transplant. 2016;20:1148-51.
- [Google Scholar]
- Schimke immune-osseous dysplasia: A case report. Saudi J Kidney Dis Transpl. 2015;26:987-91.
- [Google Scholar]
- Juvenile variant of Schimke immunoosseous dysplasia. Am J Med Genet. 1994;49:266-9.
- [Google Scholar]
- Schimke immuno-osseous dysplasia: A cell autonomous disorder? Am J Med Genet A. 2006;140:340-8.
- [Google Scholar]
- Early onset cerebral infarction in Schimke immuno-osseous dysplasia: A case report. Iran J Child Neurol. 2018;12:126-32.
- [Google Scholar]
- Internal carotid artery surgical revascularization in a pediatric patient with Schimke immuno-osseous dysplasia. J Neurosurg Pediatr. 2015;15:189-91.
- [Google Scholar]
- Rituximab resistant evans syndrome and autoimmunity in Schimke immuno-osseous dysplasia. Pediatr Rheumatol Online J. 2011;9:27.
- [Google Scholar]
- Tremor onset with acute frontal infarct and disappearance with the second stroke. Neurosciences (Riyadh). 2015;20:164-6.
- [Google Scholar]
- Tremor following ischemic stroke of the posterior thalamus. J Neurol. 2010;257:1934-6.
- [Google Scholar]




