

Translate this page into:
Alport's Syndrome: A Rare Clinical Presentation with Crescents
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Alport's syndrome (hereditary nephritis) is a familial disorder, which usually affects young males with clinical presentation of hematuric and glomerular disease. We report a rare case of Alport's syndrome in a 16-year-old male with typical extrarenal manifestations and renal biopsy findings with crescents.
Keywords
Alport's syndrome
crescents
hematuria

Introduction
Alport's syndrome is an inherited disorder and refers to the clinical triad of hereditary nephritis, sensorineural deafness, and ocular abnormalities,[1] which typically occur in the second to fourth decade. It attributes to about 1% of end-stage renal failure.[2] The most common presentation is persistent microscopic hematuria starting as early as 5 years of age, which invariably occurs in all males. Recurrent gross hematuria occurs in 40%–60% cases during infancy and early childhood.[3] Proteinuria develops later. Bilateral sensorineural hearing loss is second most common feature occurring in 55% in males and 45% in females.[4] It becomes apparent by late childhood to early adolescence in boys with X-linked disease.[5] Ocular manifestations occur in 15%–30% cases.[6] Anterior lenticonus is virtually pathognomonic of Alport's syndrome[7] and is known as “oil droplet in water” appearance.[8]
Case Report
A 16-year old, without any major past medical illness, had complaints of intermittent headache, visual blurring, vomiting since last 2 months, and periorbital puffiness since 20 days. His headache was holocranial, nonthrobbing, and associated with episodes of vomiting and transient bilateral visual blurring without any redness of eyes or ocular pain. He never developed any focal deficit, episode of seizure, or loss of consciousness. After 1.5 months of onset of these symptoms, he developed facial puffiness, which was more in the morning. There was no complaint of fever, preceding sore throat, pedal edema, decrease in urine output, dysuria, hematuria, anorexia, or any other significant complaint.
He initially visited a local clinic where he was found to have high blood pressure (170/100 mmHg). On routine tests, he was found to have a creatinine of 2.1 mg/dL and urine analysis showed Protein 2+, RBC 2-5/hpf, Pus cells 1-2/hpf, and Casts granular 6-8/lpf. He was treated with Amlodipine and referred to our hospital for further management.
At our hospital, on examination, he was conscious and oriented. His general physical examination showed pulse 92/min, blood pressure 180/106 mmHg, mild evidence of pallor, no edema, cyanosis, clubbing, or lymphadenopathy. His systemic examination revealed normal respiratory, cardiovascular, and neurological systems. His fundus examination revealed “oil drop” appearance. On investigations, he had hemoglobin 8.9 g/dL, total leucocyte count 5,700/cmm, platelets 1,77,000/cmm, blood urea nitrogen 20 mg/dL, creatinine was 2.4 mg/dL, sodium 134 meq/L, potassium 5.3 meq/L, bicarbonate 20 mmol/L, calcium 8.5 mg/dL, phosphorus 5.4 mg/dL, albumin 2.5 g/dL, and uric acid 5.7 mg/dL. Urine examination showed Protein 2+, RBC 4-6/hpf, Pus cells 2-5/hpf, and Casts granular 2-4/lpf. CXR was normal and electrocardiogram showed signs of left ventricular hypertrophy. Kidney biopsy was done.
Light microscopy showed 10 glomeruli with one globally sclerosed glomerulus. There were fibrocellular crescents in two glomeruli (22%), one circumferential, and one partial [Figure 1]. Glomerular capillary loops were unremarkable and without endocapillary hypercellularity. There was no mesangial expansion or hypercellularity. There was patchy tubular injury with cytoplasmic vacuoles and interstitial inflammation and significant tubular and interstitial fibrosis. Blood vessels showed medial thickening with duplication of internal elastic lamina. Immunofluorescence was negative. Electron microscopy was awaited, and in view of presence of crescents, autoimmune profile was sent (ANA, ANCA, anti-GBM antibody), which were negative and complement levels C3 and C4 were in normal range. Electron microscopy later showed normocellular glomeruli with flattening of foot processes. All the loops showed prominent irregularities of lamina densa with splitting and some regions showing thinning alternating with thick areas. The glomerular basement membrane (GBM) thickness was variable with one loop showing thickness of 170 nm only [Figure 2]. One region showed basket-weave appearance and there were no dense deposits or any sclerosis – diagnostic of Alport syndrome. Slit lamp examination showed bilateral anterior lenticonus (oil drop in water appearance), whereas audiometry showed bilateral mild to moderate sensorineural hearing loss.

- Light microscopy showing fibrocellular crescent
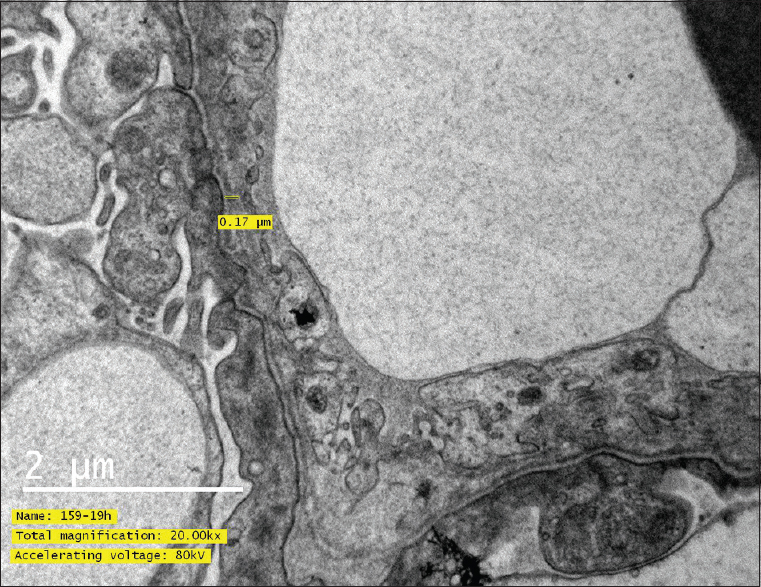
- Electron microscopy showing glomerular basement membrane thickness 170 nm
His mother and maternal siblings were also screened for occult disease. However, none of them had hematuria/proteinuria in their urine analysis. Genetic analysis was denied by the patient and family.
Discussion
Over 80% cases of Alport's syndrome are X-linked and young men are most affected. Remaining 10%–15% cases are of autosomal inheritance.[2] It is caused by mutations in basement membrane (type IV) collagen encoded by COL4A5 (X chromosome). Most other patients have autosomal recessive or dominant Alport's syndrome due to mutations in COL4A3 or COL4A4 coded by chromosome 2.[9] The 10%–15% autosomal inherited cases, after a consanguineous marriage, differ from classical XL Alport in terms of men and women being equally affected, early progression to ESRD before 20 years of age and hematuria with extrarenal manifestations being rare.[10]
Investigation of choice remains kidney biopsy. Light microscopy shows increased mesangial matrix, capillary wall thickening, occasional focal segmental glomerulosclerosis/tubulo-interstitial nephritis, or interstitial foam cells suggestive of proteinuria. Immunofluorescence is negative. Electron microscopy shows thinning, thickening, splitting, and basket weaving of the lamina densa.[11] Immunohistology for α3 (IV) and α5 (IV) with EM findings are definitive for diagnosis of Alport. Crescents in Alport's syndrome is very rare and not well established.[12] As per data till date, crescents in Alport's could just be a rare additional biopsy feature or an extension of the classical pathological appearance, suggestive of the faster progression and poor prognosis of the disease. Afonso et al. reported 20% crescents in their biopsy of Alport's syndrome,[12] which is similar as ours. Chugh et al. also reported 1 out of their 63 patients with Alport's to have crescents.[13] Chang et al. hypothesized the pathogenesis of crescent formation. They speculated that high intraglomerular capillary pressure and defective synthesis of collagen IV leads to loss of structural integrity of the GBM, thereby leading to rupture of capillary loops and formation of crescents.[14] It also highlights the importance of electron microscopy even in crescentic glomerulonephritis, as short of EM their case would have been labeled as pauci-immune glomerulonephritis. Another case report from Harris et al. mentions a rare presentation of Alport's as crescentic gleomerulonephritis with terminal renal failure in a sibling of Alport's.[15]
Alport or hereditary nephritis commonly presents with hematuria as a renal manifestation. Microscopic hematuria is persistent and invariable in males affected by Alport's disease. Hematuria may present with/without bilateral anterior lenticonus and sensori-neural hearing loss. Presence of these extrarenal signs is not prerequisite for its diagnosis, although lenticonus is a pathognomonic feature. Also, being an X linked and rarely autosomal inherited disease, patients with Alport designate a strong family history as much as in 90% cases. Heterozygote females and autosomally inherited Alport's may have intermittent microscopic hematuria. The diagnosis in our case was established from extrarenal manifestations and kidney biopsy characteristic of Alport's syndrome.
The atypia in our case was absence of clinically significant hematuria, family history, and presence of crescents in renal biopsy. Our case did not have significant hematuria despite having crescents on biopsy. There was no family history of hematuric illness and urine examination of family members was negative for hematuria. There was presence of crescents in kidney biopsy, which was unusual for Alport's disease. The presence of focal fibrocellular crescents is a rare histological finding, the significance of which is not known from literature.[12]
Presently the patient is doing well on a single antihypertensive with his blood pressure under control. As a part of supportive treatment, he has been advised hearing aids for his hearing impairment and for a regular follow-up.
Conclusion
We report a rare case of Alport's syndrome in a young adolescent male with typical extrarenal manifestations and renal biopsy findings having crescents.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Alport syndrome: Overview. In: Turner NN, Lameire N, Goldsmith DJ, Winearls CG, Himmelfarb J, Remuzzi G, eds. Oxford Textbook of Clinical Nephrology. Oxford University Press; 2015. p. :2695.
- [Google Scholar]
- Alport syndrome: An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine. 1999;78:338-60.
- [Google Scholar]
- Psychosocial impact of an X-linked hereditary disease: A study of Alport syndrome patients and family members. Child Care Health Dev. 2000;26:239-50.
- [Google Scholar]
- Alport's syndrome: A report of 58 cases and a review of the literature. Am J Med. 1981;70:493-505.
- [Google Scholar]
- Alport's syndrome: Case report and review of ocular manifestations. Ann Ophthalmol. 1977;9:1527-30.
- [Google Scholar]
- Bilateral anterior lenticonus: Scheimpflug imaging system documentation and ultrastructural confirmation of Alport syndrome in the lens capsule. Arch Ophthalmol. 2000;118:895-7.
- [Google Scholar]
- Distribution of α-chains of type IV collagen in glomerular basement membranes with ultrastructural alterations suggestive of Alport syndrome. Nephrol Dial Transplant. 2001;16:945-52.
- [Google Scholar]
- Alport syndrome–A rare histological presentation. Port J Nephrol Hypert. 2010;24:51-5.
- [Google Scholar]
- Hereditary nephritis (Alport's syndrome) – Clinicalprofile and inheritance in 28 kindreds. Nephrol Dial Transpl. 1993;8:690-5.
- [Google Scholar]
- A rare cause of necrotizing and crescentic glomerulonephritis in a young adult male. Am J Kid Dis. 2005;45:956-60.
- [Google Scholar]
- Alport's syndrome representing as crescentic glomerulonephritis: A report of two siblings. Clin Nephrol. 1978;10:245-9.
- [Google Scholar]




