

Translate this page into:
Novel Homozygous FAN1 Mutation in a Familial Case of Karyomegalic Interstitial Nephritis
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Karyomegalic interstitial nephritis (KIN) is a rare genetic kidney disease associated with a mutation in FAN1 gene and is often underdiagnosed. The histomorphology demonstrates chronic interstitial nephritis with tubular epithelial cells showing bizarre enlarged nuclei. We present a case report of a 47-year-old multiparous South-Indian woman presenting with bilateral pitting pedal oedema and mild hypertension. At the time of presentation, her serum creatinine was 1.52 mg/dL and urine analysis showed mild proteinuria. Kidney biopsy showed features of tubular injury with bizarre enlarged nuclei and focal mild chronic tubulointerstitial nephritis. Immunohistochemistry was negative for cytomegalovirus (CMV) Ag and SV40 Ag. Real-time polymerase chain reaction (PCR) done for CMV and BK virus genomes was negative. Relevant family history was that her older brother was also diagnosed with kidney failure and is on renal replacement therapy. Genetic analysis for FAN1 gene of the proband and her sibling showed two rare mutations of the FAN1 gene in the exon 4, of which, one is non-synonymous mutation and the other is a stop-gain mutation in the proband. This case illustrates a rare presentation of karyomegalic interstitial nephritis in siblings with previous unknown FAN1 gene mutations.
Keywords
FAN1 mutation
karyomegay
interstitial nephritis

Introduction
Karyomegalic interstitial nephritis (KIN) is a rare genetic kidney disease that was first reported as dysplastic changes in the renal epithelium in a woman suffering from hepatocellular carcinoma by Burry et al. in 1974.[1] Later, the acquired cases of KIN due to toxins and viruses have been reported.[2] A case of KIN in 22-year-old male suffering from relapsed Hodgkin's lymphoma on treatment with ifosfamide was reported by Jayasurya et al.[3] Another case by McCulloch et al. reported three adolescent patients who recovered from their initial treatment for Ewing's sarcoma but developed a KIN attributed to ifosfamide therapy.[4] The other etiologic factors identified are heavy metals and mycotoxins, particularly ochratoxin A.[5] One case report of KIN associated with focal segmental glomerulosclerosis (FSGS) has been described by Radha. et al.[6] The natural course of the disease is either nonprogressive or progression to end-stage kidney disease requiring renal replacement therapy. Recurrence of KIN in the allograft kidney is also reported.[7] The genetic association is most commonly seen with FAN1 gene mutations in exon 12. So far, to our knowledge, 42 cases of KIN have been reported with some showing FAN1 gene mutations with cytogenetic location 15q13.3.
We report a middle-aged multiparous lady with KIN with two rare mutations of the FAN1 gene in the exon 4, of which, one is nonsynonymous mutation and the other is a stop-gain mutation.
Case Report
A 47-year-old multiparous, South-Indian woman was referred to the department of nephrology with the complaints of bilateral pitting pedal oedema and mild hypertension. She gave a history of steroid intake for 3 months in view of musculoskeletal inflammation of lower limbs. She is hypertensive on cilnidipine 5 mg OD with no other known comorbidities. There was no previous history of exposure to ifosfamide, ochratoxin A or intake of alternative medicine. Family history revealed ongoing kidney failure in one of her two brothers who is currently on continuous ambulatory peritoneal dialysis. Her brother's primary kidney disease is not known.
On physical examination, BP of 130/80 mm Hg, mild pallor and bilateral mild pitting pedal oedema were detected. Blood investigations showed Hb = 10 gm/dL (11–14 g/dL), serum creatinine = 1.52 mg/dL (0.5–1 mg/dL) and blood urea = 46 mg/dL (7–20 mg/dL). Fasting lipid profile revealed cholesterol = 232 mg/dL (less than 200 mg/dL), triglycerides = 168 mg/dL (less than 150 mg/dL), HDL = 52 mg/dL (above 60 mg/dL) and LDL = 157 mg/dL (less than 100 mg/dL). Urinalysis showed trace proteinuria. 2D-Echo revealed mild tricuspid regurgitation, mild pulmonary arterial hypertension and an ejection fraction of 64%. The thyroid function test and serum electrolyte values were normal. A percutaneous kidney biopsy was performed on the left side. The renal tissue was examined under light microscopy using Hematoxylin and Eosin (H and E), periodic acid Schiff (PAS), periodic silver methenamine (PASM) and Masson's trichrome stain. Five of 28 glomeruli were globally sclerosed while the remaining glomeruli showed patent capillary loops with normal mesangial cellularity. There was no evidence of diffuse thickening of glomerular capillary loops, segmental sclerosis, endocapillary or extracapillary proliferation. The tubules showed tubular injury in the form of loss of brush borders and flattening of epithelium. Many proximal and distal convoluted tubules showed bizarre enlarged nuclei with prominent nucleoli [Figure 1]. The interstitium showed mild lymphocytic infiltrate and focal mild fibrosis. No granuloma was seen. The interstitial fibrosis and tubular atrophy accounted for about 5% of the renal cortex. The interlobular arteries showed no specific lesion. Few arterioles showed similar bizarre enlarged nuclei. Immunohistochemistry staining for cytomegalovirus antigen and simian virus 40 antigen were negative. Immunofluorescence staining for IgG, IgM, IgA, C3, C1q, kappa and lambda light chains were negative. Real-time PCR for CMV and BK virus genomes were negative.
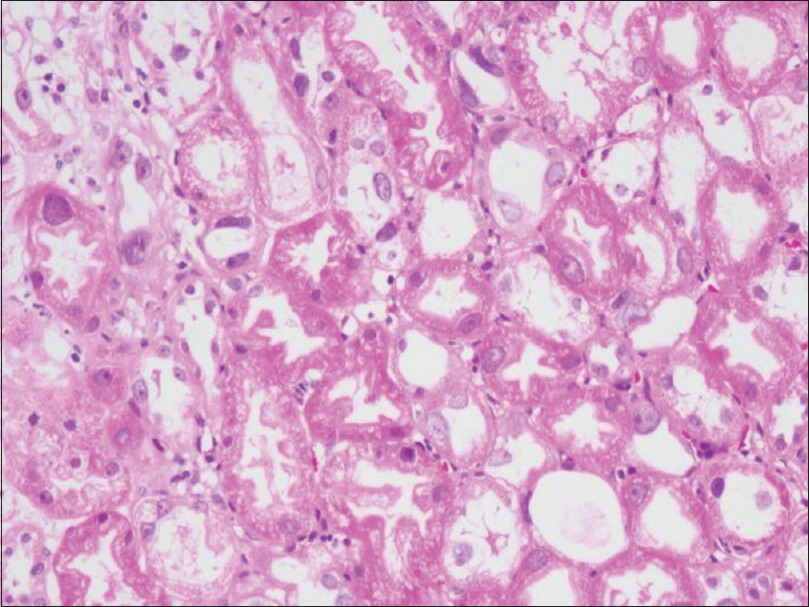
- Tubular epithelial cells showing karyomegalic nuclei (H and E 200X)
Genetic analysis of the proband and her affected male sibling was done with 5 ml of the peripheral blood sample from both, and DNA was isolated. A total of 180 healthy ethnically matched controls were also included in the present study. Primers were designed to amplify all the amino-acid coding regions (exon 2-14) of FAN1 spanning exon-intron boundaries, using Primer-3 software, version 0.4.0 (http://bioinfo.ut.ee/primer3-0.4.0). Exon 2 was amplified in three separate fragments (exon 2A, 2B and 2C).
Each PCR was carried out in 10.0μl reaction mixture using 2X EmeraldAmp GT PCR master mix® (Takara Bio INC, Japan), 0.1 pM of each forward and reverse primers, 20.0 ng genomic DNA. Direct bidirectional sequencing of the amplicons was carried out using an ABI3700 DNA analyser. Sequences were assembled with reference sequences using AutoAssembler software; version 1.4.0 (Applied Biosystem, Foster City, CA, USA), and the mutations were noted.
Genetic analysis identified two rare, deleterious mutations in the exon 4 of which one is nonsynonymous mutation; rs745320524 (c.1356T>G, p.Asn452Lys) and the other is a stop-gain (loss of function) mutation; rs746677293 (c.1369C>T, p.Gln457X). Interestingly, both the mutations were in homozygous condition and are in very close proximity [Figure 2]. Both the mutations were also observed in the proband's affected sibling.
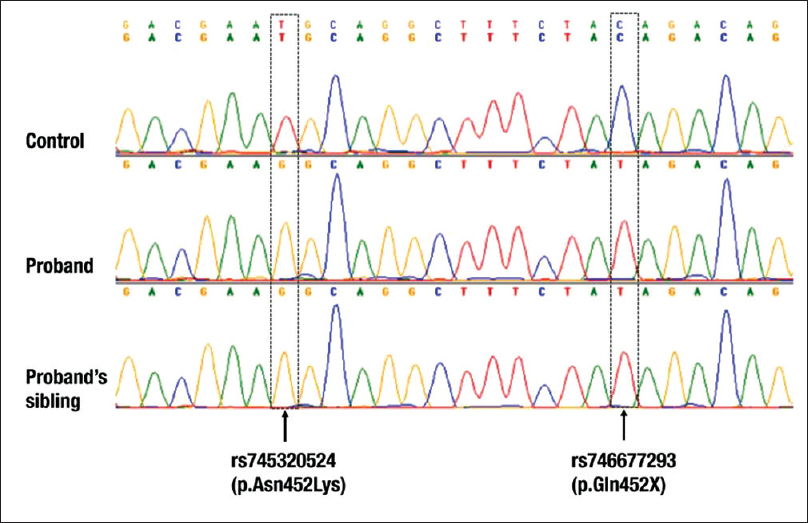
- Electropherogram of the proband and her older brother along with a control, showing FAN1 homozygous mutations in exon 4
The mutation rs746677293 (p.Gln457X) is a loss of function mutation with minor allele frequency (MAF) <0.001 (rare mutation). Because the mutation is in homozygous condition, the protein will be truncated midway, at 457th amino acid. Hence, a fully functional FAN1 protein (1017 amino acids) is absent in the patient and is a possible cause of the observed phenotype. Treatment was initiated with oral prednisolone 30 mg daily. It was tapered and discontinued when the serum creatinine fell to 1.4 mg/dl after 10 weeks. Optimal blood pressure was obtained using amlodipine 5 mg OD and chlorthalidone 6.25 mg OD. Post initiation of therapy, urinalysis was normal with nil proteins in urine.
Discussion
KIN is a rare disorder with less than 50 cases being described in medical literature to date.[8] It was first reported as dysplastic changes in the renal epithelium in a 22-year-old woman suffering from hepatocellular carcinoma by Burry et al., 45 years ago.[1] The term ‘KIN’ was then introduced by Mihatsch et al. in 1979.[2] Initially thought to be hereditary, KIN was later associated with an autosomal recessive mutation in FAN1 gene which encodes Fanconi anaemia associated nuclease 1. FAN1 gene is involved in DNA interstrand cross-link repair and also encodes protein with 5’ flap endonuclease and 5’-3’ exonuclease activity. FAN1 gene is expressed ubiquitously in skin (RPKM 6,7), kidney, neuronal tissue (RPKM 6,7) and over 25 other tissues.[910] KIN is of multifactorial etiology, including hereditary, acquired and idiopathic causes.[8] The patient usually presents in the third or fourth decade though rarely reported in children.[7] They present with impaired kidney function, minimal proteinuria, glycosuria seen in >75%, and less than one-third presenting with urinary sediment abnormalities, mostly commonly haematuria.[11] Extra-renal manifestations are uncommon. Repeat kidney biopsy specimen usually showing non-specific but severe chronic interstitial fibrosis and tubular changes, associated with nonspecific glomerulosclerosis and vascular lesions. Hallmark features of the disease include presence of karyomegalic tubular epithelial cells, lining the proximal and distal tubules, characterized by markedly enlarged and hyperchromatic nuclei.[12] Studies have suggested a degenerative cellular process, as evidenced by a low index of Ki-67 staining in renal tubular epithelial cells.[7] Urine analysis as a diagnostic aid is uncertain but its potential benefits as a screening tool could be used to identify high-risk individuals such as those with a positive family history of KIN and kidney failure.[7]
In this case report, the patient was not exposed to nephrotoxins nor had any malignancy. Kidney biopsy revealed typical features of KIN. Genetic analysis disclosed a riveting finding of two rare mutations of the FAN1 gene in the exon 4 of which one is non-synonymous mutation and the other is a stop-gain mutation not noted earlier, in the proband as well as her CKD affected sibling. The patient is being regularly followed up.
Conclusion
We present a rare case of KIN who presented with bilateral pitting pedal oedema, mild hypertension, deranged kidney function and minimal proteinuria. Genetic analysis of the patient and her affected sibling showed two rare mutations of the FAN1 gene in the exon 4 which has not been described in literature earlier.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Extreme dysplasla in renal epithelium of a young woman dying from hepatocarcinoma. J Pathol. 1974;113:147-50.
- [Google Scholar]
- Systemic karyomegaly associated with chronic interstitial nephritis. A new disease entity? Clin Nephrol. 1979;12:54-62.
- [Google Scholar]
- Karyomegalic interstitial nephropathy following ifosfamide therapy. Indian J Nephrol. 2016;26:294-7.
- [Google Scholar]
- Karyomegalic-like nephropathy, Ewing's sarcoma and ifosfamide therapy. Pediatr Nephrol. 2011;26:1163-6.
- [Google Scholar]
- Karyomegaly of tubular kidney cells in human chronic interstitial nephropathy in Tunisia: Respective role of Ochratoxin A and possible genetic predisposition. Hum Exp Toxicol. 2004;23:339-46.
- [Google Scholar]
- Karyomegalic interstitial nephritis with focal segmental glomerulosclerosis: A rare association. Indian J Nephrol. 2014;24:117-9.
- [Google Scholar]
- Karyomegalic interstitial nephritis in a renal allograft. Am J Transplant. 2018;19:285-90.
- [Google Scholar]
- Karyomegalic interstitial nephritis: A case report and review of the literature. Medicine. 2016;95:e3349.
- [Google Scholar]
- FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. 2012;44:910-5.
- [Google Scholar]
- FAN1 FANCD2 and FANCI associated nuclease 1 [Homo sapiens (human)]-Gene-NCBI [Internet] Ncbinlmnihgov 2019. Available from: https://wwwncbinlmnihgov/geneDb=gene&Cmd=DetailsSearch&Term=22909
- [Google Scholar]
- Karyomegalic interstitial nephritis: Report of 3 new cases and review of the literature. Clin Nephrol. 2006;65:349-55.
- [Google Scholar]
- Karyomegalic nephropathy: An uncommon cause of progressive renal failure. Nephrol Dial Transplant. 2002;17:1914-20.
- [Google Scholar]




