

Translate this page into:
Transformation of membranous into anti-GBM nephritis
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Membranous nephropathy is a common glomerular disease. We report a 50-year-old man with a history of membranous nephropathy in remission, who presented with acute kidney injury, proteinuria, hematuria, and hypertension. He also had a high anti-glomerular basement membrane (anti-GBM) antibody titer and crescent transformation of primary pathology. The kidney functions deteriorated rapidly despite aggressive therapy with cyclophosphamide, methylprednisolone, and plasmapheresis.
Keywords
Anti-glomerular basement membrane nephritis
crescentic glomerulonephritis
membranous nephropathy
schistosomal glomerulopathy

Introduction
Membranous nephropathy accounts for about one-third of cases of adult glomerular diseases[1] and is one of the leading causes of end-stage renal disease (ESRD). Despite its favorable prognosis, due to its inherent tendency of spontaneous remission in up to 30% of patients,[2] the number of deaths due to membranous disease is high due to increasing number of patients with membranous nephropathy.[3]
Crescentic transformation of membranous nephropathy is rare, and may be associated with anti-glomerular basement membrane (anti-GBM) disease[4] ANCA-associated crescentic transformation,[5] or may be without any association.[6] If not diagnosed early and treated aggressively, it might result in ESRD or death.[7]
The etiology and pathogenesis of the transformation are poorly understood, and most of the observations are hypothetical.
Case Report
A 50-year-old man was referred to our hospital 3 years ago with a history of bloody diarrhea along with central abdominal pain of colicky nature. The clinical examination was normal except for pallor and diffuse tenderness in the left flank. Investigations showed a hemoglobin of 16.3 g/ dL, WBC 10,700 (neutrophil count 51.9%, lymphocytes 22.5%, monocytes 8.2%, eosinophil 16.95%, basophil 0.5%), platelets 1,70,000/mm3, ESR 101 mm/h, blood sugar 90 mg/dL, urea 15.5 mg/ dL, creatinine 0.70 mg/dL, Na 136 meq/L, K 4.8 meql/L, and Cl 104 meq/L. Urinalysis showed a pH 5.8, protein +1, blood +3, Hb +3, RBCs 30, and no WBCs. Serology showed hepatitis HBsAg negative, hepatitis C antibody nonreactive, and schistosomal antibody titer 1:4196.
The patient had undergone a colonoscopy at a private hospital 1 week prior to visiting us, which reported the presence of schistosomal ova along with multiple polyps. Another colonoscopy at our hospital also gave the same results. Two of the polyps were resected and histopathology report confirmed the presence of tubulovillous adenoma with no evidence of malignancy. He was treated with oral praziquantel.
A month later he was readmitted with pain in the left lumber region, which was dull and persistent in nature, with no history of blood in stool and no change in the color of urine. An examination showed a palpable left kidney with tenderness on deep palpation along with bilateral pedal edema. A kidney ultrasound also showed enlargement of both kidneys with no hydronephrosis. A computerized tomographic (CT) scan of the abdomen revealed left renal vein thrombosis and other laboratory findings were essentially as before.
His serology was done which showed ANA and Anti-ds DNA was negative; complement C3 1.6, C4 0.588, HepBsAg, and hepatitis C antibody were negative; factor V Leiden mutation negative; prothrombin mutation negative; protein C 75 (70- 140), protein S 55 (65- 140), anti-thrombin III 61 (80-120). VDRL negative, rheumatoid factor negative, anti-CCP antibody negative, Smith antibody negative, Sjögren A negative, and Sjögren B negative. Twenty-four hours urinary protein was 14 g.
Kidney biopsy showed 10 glomeruli, one of which was completely sclerosed, the remaining showed rigidity of the capillary wall [Figure 1]. There was no crescent formation or fibrinoid necrosis. Tubules and vessels showed no significant pathology, while interstitium showed mild mononuclear inflammatory cell infiltration and there was no significant fibrosis. Immunofluorescence study showed eight glomeruli with +3 granular membrane positivity with IgG and C3 and also there was focal 1+ positivity with IgM, while fibrinogen and IgA were negative. Electron microscopy showed subepithelial dense deposits [Figure 2]. All the results confirmed the diagnosis of membranous nephropathy.
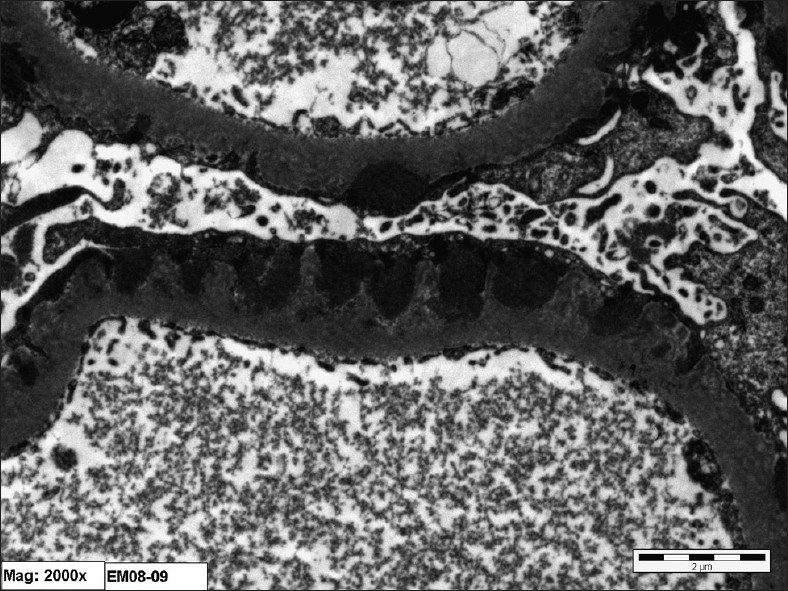
- Electron photomicrograph showing diffuse fusion of epithelial foot processes and wide spread subepithelial dense deposit

- Section of a glomerulus with a minimally thickened basement membrane (PAS stain, ×40)
Oral anticoagulant, prednisolone 30 mg with cyclosporin 150 mg twice a day was started. A follow-up colonoscopy was performed twice each 6 months apart that revealed adenomatous polyps with no evidence of malignancy. On follow-up after 6 months, he was stable with mild pedal edema, urea 35 mg/dL, creatinine 0.82 mg/dL, Na 139 meq/L, K 4.9 meq/L. Urinalysis showed protein +2, RBC nil, and 24-h urinary protein was 1.09 g, Cyclosporin was reduced to 50 mg BID and prednisolone to 5 mg OD.
On 1-year follow-up, the patient was found to be in complete remission, urea 18.6 mg/dL, creatinine 1.04 mg/ dL, Na132 meq/L, K 5.0 meq/L, and 24-h urinary protein 0.06 g; therefore, cyclosporine and steroids were discontinued.
After 6 months of stopping all medications, he was stable, and urinary sediments showed no proteinuria or hematuria, and kidney functions were normal.
Two weeks after the last follow-up that was 2½ years after his first presentation, he was readmitted with a history of left lumbar pain and dark brown urine for 3 days. Examination showed diffused tenderness in the left lumbar region, blood pressure of 140/80, and pedal edema. On investigation, urinalysis showed blood 3+, protein 2+ with no cast, 24-h urinary protein was 5.0 g, urea 34 mg/dL, creatinine 3.42 mg/dL, Na 135 meq/L, K 5.5 meq/L, ANA negative, anti-DNA negative, and C3 and C4 normal cANCA negative.
Ultrasound and Doppler of the kidney were performed to rule out renal vein thrombosis, which revealed a normal size kidney and there was no evidence of renal vein thrombosis. He was started on oral prednisolone 60 mgs once a day.
A kidney biopsy showed 24 glomeruli, of which three were globally obsolescent, seven showed cellular crescents, one showed fibrocellular crescent, two glomeruli showed segmental endocapillary proliferation, four showed fibrinoid necrosis in glomerular crescents [Figures 1 and 3] in the glomerular tufts. Silver stain showed moth-eaten appearance and spike formation. Approximately 20% of the specimen showed tubular atrophy and interstitial fibrosis. There was no vasculitis, thrombosis, or fibrinoid necrosis of blood vessels. The Congo red stain was negative.

- A cellular crescent in one glomerulus (methenamine silver Jones stain, ×40)
Immunofluorescence microscopic study showed diffuse granular capillary positivity with IgG 3+ and C3 1+, while IgA, IgM, C1q, and albumin were negative [Figure 4]. Electron microscopy examination showed diffuse effacement of the visceral epithelial cell foot process with widespread microvilli degeneration of the glomerular epithelial cell. The mesangium showed mild focal increase in mesangial matrix and cellularity; subendothelial or mesangial dense deposits were not seen [Figure 4].

- Immunofluorescence study for IgG showing granular diffuse capillary staining (2+)
Serology showed very high titer of anti-GBM antibodies, 190 u/mL. The patient was shifted to intravenous methylprednisolone and pulse cyclophosphamide and plasmapheresis were started. He showed progressive rise in serum creatinine with gradual decline in urinary output: hemodialysis was started due to volume overload and an impending pulmonary edema. Treatment continued on plasmapheresis, which was stopped after six sessions; pulse cyclophosphamide was given for 3 months and later on stopped due to persistent anuria.
He was put on maintenance hemodialysis three times a week. During hemodialysis, he was followed with a monthly titer of anti-GBM antibody that remained high for 4 months and gradually declined and disappeared after 6 months from the onset of anti-GBM disease. The schistosomal antibody titer was also followed which declined very slowly and the last titer was 1:1024, performed after three months of starting hemodialysis.
Discussion
The transformation of membranous nephropathy into crescentic glomerulonephritis is rare but well-known phenomena and has significant morbidity and mortality if not diagnosed early and treated promptly. Our case had some unique features: on his first presentation, the patient had a history of schistosomal infestation, had renal vein thrombosis, responded promptly, and sustained remission after the treatment with steroid and cyclosporine; later on when he presented for the second time with acute kidney injury, the renal biopsy showed granular deposits in immunofluorescence rather than linear deposits with positive serum anti-GBM antibody.
Membranous nephropathy is not a classical lesion of schistosomiasis and schistosomal infestation mostly associated with mesangial proliferation, membranoproliferative glomerulonephritis, and focal segmental glomerulosclerosis. It is seldom described in association with mansoni schistosomiasis.[8] The prognosis of established schistosomal glomerulopathy is poor and the disease usually progresses to ESRD despite the eradication of schistosomal infection, whereas[9] the response of the membranous disease in our patient was good and sustained. It is difficult to say whether schistosomiasis was the cause of the membranous disease or it was only coincidently associated,[10] but the disease behaved like an idiopathic membranous disease.
This case also shows that transformation of the membranous disease into crescentic nephritis along with positive anti-GBM antibody could be associated with diffuse granular deposits rather than linear deposits on immunofluorescence, which suggests the possibility of release of the GBM antigen during the membranous disease contributing to the development of anti-GBM nephritis. This feature is being reported in the literature with the possibility of concomitant immune-mediated mechanism.[11]
The reason why membranous nephropathy has predilection for transformation to anti-GBM antibody nephritis and how this transformation occurs are not clear. Therefore, it is still a speculation that it might be due to membrane deposits that alter the GBM and cause the release of normal or endogenous GBM antigen into the circulation, which results in the formation of anti-GBM antibody,[12] or due to alteration and release of “hidden” GBM antigen which results in the formation of anti-GBM antibody.[13]
Recent advancement in understanding the etiology and pathogenesis of membranous disease and anti-GBM nephritis revealed many facts, especially the identification of M-type phospholipase A2 receptor antigen as an etiological factor of membranous nephropathy,[14] and autoimmune nature of good pasture or anti-GBM nephritis caused by conformational change in the structure of basement membrane, that is, “conformopathy” of the membrane by an autoimmune phenomena.[15] It might be the release of M-type phospholipase A2 receptor antigen that causes a “conformational” change in the GBM that precipitates anti-GBM nephritis.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Changing etiologies of unexplained adult nephrotic syndrome, a comparison of renal biopsies findings from 1976-1979 and 1995-1997. Am J Kidney Dis. 1997;30:621-31.
- [Google Scholar]
- Prognosis of untreated patients with idiopathic membranous nephropathy. N Eng J Med. 1993;329:84-9.
- [Google Scholar]
- Distribution of primary renal disease leading to end-stage renal failure in United States, Europe and Australia/New Zeeland. Results from an international comparison study. Am J Kidney Dis. 2000;35:157-65.
- [Google Scholar]
- Acute renal failure in membranous glomerulonephropathy: A result of superimposed crescentic glomerulonephritis. J Am Soc Nephrol. 1995;6:1541-6.
- [Google Scholar]
- Crescentic transformation in primary membranous glomeulopathy: association with anti-GBM antibody. Saudi J Kidney Dis Transplant. 2007;18:599-602.
- [Google Scholar]
- Membranous glomerulonephritis with ANCA-associated necrotizing and crescentic glomerulonephritis. Clin J Am Soc Nephrol. 2009;4:299-308.
- [Google Scholar]
- Crescentic transformation in primary membranous glomerulonephritis. Postgrad Med J. 1991;67:546-6.
- [Google Scholar]
- Characterisation of kidney lesions in early schistosomal-specific nephropathy. Nephrol Dial Transplant. 1988;3:392-8.
- [Google Scholar]
- Schistosomial mansoni associated glomerulopathy. Rev Inst Med Trop Sao Paulo. 1999;41:269-72.
- [Google Scholar]
- Anti-GBM- associated crescentic glomerulonephritis with discreate IgG deposition, but with no electron-dense materials in glomeruli. Nephrol Dial Transplant. 1996;11:2070-3.
- [Google Scholar]
- Evolution of membranous nephropathy into anti-glomerular basement membrane glomerulonephritis. N Engl J Med. 1974;290:1340-4.
- [Google Scholar]
- Transformation of membranous glomerulonephritis into cresentic glomerulonephritis with glomerular basement membrane antibodies. Serial determinations of anti-GBM before the transformation. Nephron. 1984;38:134-7.
- [Google Scholar]
- M-type phospholipase A2 receptor as a target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11-21.
- [Google Scholar]
- Molecular architecture of the Goodpasture autoantigen in anti GBM nephritis. N Engl J Med. 2010;363:343-54.
- [Google Scholar]




