

Translate this page into:
Gitelman's syndrome: Rare presentation with growth retardation
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Gitelman's syndrome is an autosomal recessive disorder characterized by hypokalemic metabolic alkalosis, hypokalemia, hypomagnesaemia, hypocalciuria, hyperreninemia and without hypertension. Gitelman's syndrome is caused by mutations of the SLC12A3 gene, which encodes the Na/Cl co-transporter (NCCT) in the distal convoluted tubule. Majority of cases manifest during adolescence or adulthood and growth retardation is not the common feature. We report a rare presentation of Gitelman's syndrome in a four-year-old boy with growth retardation.
Keywords
Gitelman's syndrome
hypocalciuria
hypokalemia
hypomagnesaemia
metabolic alkalosis

Introduction
Gitelman's syndrome (GS) was first described by Gitelman et al. in 1966.[1] It is an autosomal recessive salt-losing renal tubulopathy that is characterized by hypokalemic metabolic alkalosis, hypomagnesaemia, hypocalciuria, and secondary aldosteronism.[1234] It is a rare disease and its clinical symptoms may include fatigue, cramp, muscle weakness, carpopedal spasms, and rarely may include serious symptoms such as paralysis and sudden cardiac arrest.[5] Growth retardation can be seen with GS, but not frequent as in other tubular disorders like Bartter syndrome (BS).[67] Gitelman's syndrome is caused by mutations of the SLC12A3 gene, which encodes the Na/Cl co-transporter (NCCT) in the distal convoluted tubule.[23] The diagnosis of GS is based on clinical symptoms and biochemical abnormalities.
Case Report
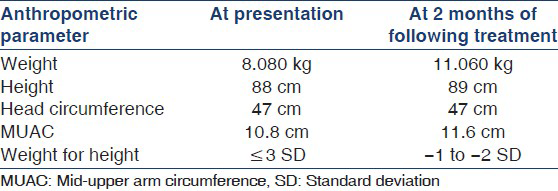
A 4-year-old child, product of non-consanguineous marriage, was brought to the Pediatric Intensive Care Unit (PICU) of Medical College Hospital with the complaints of fever, loose stools, labored breathing, weakness of all four limbs, and not growing well. There was no history of recent vaccination, diuretic intake, vomiting, seizure, headache, and chest pain. No other family member had similar illness. On examination, patient was lethargic, severely dehydrated, heart rate 126/min, respiratory rate 52/min, and blood pressure 98/64 mmHg (95th percentile for age and height 106/66). Cardiovascular examination was suggestive of sinus tachycardia. Abdomen was distended without organomegaly with diminished bowel sounds. On CNS examination, tone and power were decreased in all limbs, deep tendon reflexes were diminished, bilateral planters were flexor, and no sign of meningitis. At the time of presentation, his weight was 8.08 kg (<3rd percentile) and height was 88 cm (<3rd percentile); head circumference was 47 cm, mid-upper arm circumference (MUAC) was 10.8 cm, and weight for height ≤3 SD. Laboratory investigations showed that his hemoglobin was 9 g/dl; the total leukocyte count was 5.4 × 1000 cells per mm3 and blood and urine culture were sterile. His serum sodium was 134.8 meq/l, potassium 2.0 meq/l, calcium 7.7 mg/dl, and magnesium 1.2 mg/dl (normal 1.7-2.2 mg/dl); urea 42 mg/dl, creatinine 0.77 mg/dl, and random blood sugar was 94 mg/dl. With treatment, his hydration status improved; but hypokalemia was persisting. This was further investigated by 24-h urine analysis which showed the potassium to be 16.8 mmol/24 h (normal 2.5-125 mmol/24 h), chloride 50.40 mmol/24 h (normal 15-40 mmol/24 h), calcium 2.5 mg/24 h (normal 100-300 mg/24 h), and magnesium 1.0 mmol/24 h (normal 0.04-1.40 mmol/24 h). Arterial blood gas analysis showed metabolic alkalosis (pH 7.53, HCO3 36.3, PaCO2 46.5, and base excess +13). The urine output was 3 ml/kg/h. Renal ultrasonography showed no evidence of nephrocalcinosis. Biochemical workup showed hypokalemic metabolic alkalosis, hypomagnesaemia with hypocalciuria. Before receiving the complete reports of blood biochemistry and urinary electrolyte, we provisionally diagnosed the case as Bartter's syndrome because hypokalemic metabolic alkalosis in a four-year-old child with growth retardation is the typical presentation of BS. But the hypomagnesemia and hypocalciuria in the case were characteristics of Gitelman's syndrome. Patient was treated with oral magnesium and potassium supplementation. Patient symptoms resolved quickly as the treatment continued. He was discharged with advice to continue oral potassium and magnesium supplements. He recovered with normokalemia and remarkable weight gain at 2 months of follow-up as is being shown in the Table 1.
Discussion
Inherited salt-losing nephropathies Gitelman's syndrome and Bartter's syndrome are rare disorders. Gitelman's syndrome is having the prevalence of 1:40,000, whereas the BS is having the prevalence of one per million in the western population.[8] Current literature is limited of Indian data regarding the prevalence of these disorders. One of the largest case series of BS reported from India has the mean age of 6.5 ± 4.9 months.[8] Different case reports of Gitelman's syndrome from India are suggestive of presentation of GS in 4-6th decades of life.[910111213] Though the Gitelman's syndrome is a inherited disorder, even then the extensive search of literature could not find the reported case of Gitelman's syndrome in childhood. Perhaps present case is the first ever reported case of Gitelman's syndrome is a 4-year-old child. Symptoms of Gitelman's syndrome reported in literature range from asymptomatic to mild symptoms of cramps and fatigue to severe manifestations such as tetany, paralysis, and rhabdomyolysis.[5] Bartter syndrome presents in young children with failure to thrive vomiting, polyuria, dehydration, fever, and respiratory distress. GS is distinguished from BS by hypomagnesemia and hypocalciuria.[4]
Gitelman's syndrome results from a defect in the Na/Cl co-transporter (NCCT) and impaired salt transport in the distal convoluted tubule. As NCCT is the target of thiazide diuretics, manifestations of Gitelman's syndrome are mimicked by chronic thiazide administration. Only about 5-10% of the filtered salt load is reabsorbed in the distal convoluted tubule and compensatory responses of the thick ascending loop of henle (TALH) and the collecting tubule can nearly compensate for this deficit. Thus, patients suffering from Gitelman's syndrome exhibit only mild renal salt wasting, but downstream compensatory sodium and chloride reabsorption in the collecting duct, under the influence of increased aldosterone drives potassium secretion in this nephron segment, accounting for marked hypokalemia. Compensatory salt reabsorption in TALH enhances the luminal electropositive potential and promotes secondary calcium reabsorption. The resultant hypocalciuria is an important clue for distinguishing Gitelman's syndrome from Bartter syndrome.[57] The mechanism causing hypomagnesemia in Gitelman's syndrome is still not well understood because only 5-10% of filtered magnesium is reabsorbed in the distal convoluted tubule. Salt wasting is usually mild in Gitelman's syndrome and NSAID therapy is not usually required. Hypokalemia can often be corrected with oral potassium supplements (2-3 meq/kg/day), but some patients may also require the potassium-sparing diuretics. Oral magnesium supplements may be essential to prevent muscle cramping. Correction of hypomagnesemia may diminish the impact of renal potassium wasting and correct functional hypoparathyroidism.[14] Early recognition of this syndrome might provide normal growth development and prevent possible serious complications such as paralysis and sudden cardiac arrest. The case in this discussion was also having associated growth retardation and severe acute malnutrition and by treating the underlying condition, he responded very well with remarkable weight gain following therapy [Table 1]. This experience also emphasizes that in India, where malnutrition prevalence is very high, we should keep a very high index of suspicion of other rare causes of malnutrition in case of non-response to standard management protocol.
Source of Support: Nil
Conflict of Interest: None declared.
References
- A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221-35.
- [Google Scholar]
- Gitelman's variant of bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive na-cl cotransporter. Nat Genet. 1996;12:24-30.
- [Google Scholar]
- Identification of fifteen novel mutations in the slc12a3 gene encoding the na-cl co-transporter in Italian patients with gitelman syndrome. Hum Mutat. 2002;20:78.
- [Google Scholar]
- Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and gitelman syndromes. J Pediatr. 1992;120:38-43.
- [Google Scholar]
- Gitelman's syndrome revisited: An evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59:710-7.
- [Google Scholar]
- The biochemical diagnosis of gitelman disease and the definition of “hypocalciuria”. Pediatr Nephrol. 2003;18:409-11.
- [Google Scholar]
- Inactivation of the Na-Cl co-transporter (NCC) gene is associated with high BMD through both renal and bone mechanisms: Analysis of patients with gitelman syndrome and Ncc null mice. J Bone Miner Res. 2005;20:799-808.
- [Google Scholar]
- Childhood bartter's syndrome: An Indian case series. Indian J Nephrol. 2010;20:207-10.
- [Google Scholar]
- Gitelman's syndrome presenting as hypokalemic periodic paralysis. The Internet Journal of Endocrinology. 2008;4:3.
- [Google Scholar]
- Gitelman's syndrome presenting as recurrent paralytic ileus due to chronic renal tubular k+ wasting. J Assoc Physicians India. 2010;58:322-4.
- [Google Scholar]
- Diuretic loading test and use of bartter's normogram in diagnosing a case of gitelman's syndrome: Relook into pathophysiology. Indian J Nephrol. 2011;21:289-92.
- [Google Scholar]




