

Translate this page into:
Clinical and Genetic Profile of Indian Children with Primary Hyperoxaluria
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Primary hyperoxaluria (PH) has heterogeneous renal manifestations in infants and children. This often leads to delay in diagnosis. In the past 3 years, genetic samples were sent for seven children with a clinical diagnosis of PH. Their medical records were reviewed for clinical presentation and outcomes. Of the seven children, three were males. The median age of presentation was 4.9 years with the youngest presenting at 3 months of age. Nephrolithiasis, the most common presentation was associated with renal dysfunction in two children. Two children with no significant history presented in end-stage renal disease (ESRD). The sibling of one of the children in ESRD, with a history of consanguinity in parents, was screened for asymptomatic nephrolithiasis. Bilateral multiple renal calculi were found in majority of children followed by echogenic kidneys on ultrasound examination. Genetic analysis suggested PH Type 1 in five children and type 2 in two children. The mutations detected in our cohort were different from the previously reported common mutations. There was no obvious genotype-phenotype correlation noticed. Three children in ESRD are on maintenance dialysis. Nephrolithiasis being a common presentation of PH needs prompt evaluation. Mutations are generally population specific, and whole gene sequence analysis is critical in diagnosis.
Keywords
Nephrocalcinosis
nephrolithiasis
primary hyperoxaluria

Introduction
Primary hyperoxaluria (PH) is characterized by oxalate overproduction and elevated excretion. Clinical manifestations of PH are heterogeneous with respect to age clinical presentation, severity, and rate of progression of renal insufficiency. Three genetic forms of the disease are known till date.
PH1 is caused due to mutations in the alanine-glyoxylate aminotransferase (AGXT) gene that codes for the enzyme AGXT. It is the most severe form, accounting for 80% of all the cases.[1] Although PH 1 can present as infantile oxalosis,[2] the most common presentation is recurrent urolithiasis with nephrocalcinosis resulting in end-stage renal disease (ESRD) by the second decade of life.[3] PH 2, generally less severe than PH 1, has a similar age of presentation. It is caused by a deficiency of glyoxylate reductase/hydroxypyruvate reductase.[4] 4-hydroxy-2-oxoglutarate aldolase (HOGA1) which encodes the enzyme HOGA is mutated in PH 3.[5] Being least severe, it usually presents in the first decade of life with the less active stone formation and preserved renal function.
The confirmatory diagnosis of PH mandates genetic analysis. Genetic analysis is invaluable in deciding the appropriate management, prognostication, prenatal diagnostic testing, and sibling screening.
We report a series of seven children, five of them diagnosed with PH 1 and two with PH 2 with the aid of genetic analysis. There are no previous published case reports on PH 2 from India. The mutations seen in our children are different from those previously reported as common both within and outside the subcontinent.
Case Report
Among 211 newly diagnosed chronic kidney disease (CKD) patients over 3 years, genetic work up for hyperoxalosis was indicated in seven children.
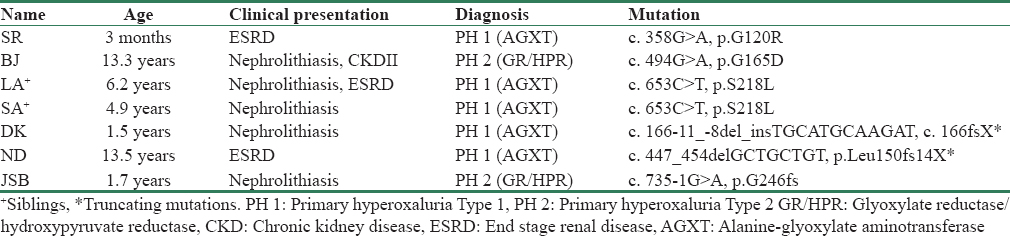
Of the seven children, three were males. The median age of presentation was 4.9 years; the youngest and the oldest child being 3 months and 13½ years, respectively. Two children were siblings born to a consanguineous couple, the younger one though asymptomatic was found to have bilateral renal calculi on screening. Five children had bilateral nephrolithiasis, renal dysfunction being evident in two of them. The other two children presented in ESRD, one among them had infantile oxalosis.
Stone analysis showed predominantly calcium oxalate monohydrate. The urinary oxalate levels of the three children with preserved renal function were elevated, mean value being 3.2 mmol/1.73 m2/day. Urine glycerate levels were not checked.
The blood samples of these children were sent to the Mayo Clinic, USA and polymerase chain reaction technique was employed for the genetic analysis. Genetic analysis confirmed PH 1 in five children and PH 2 in two children. No child had PH 3. Five children were homozygous for nontruncating mutations, and the other two had truncating mutations. No two children except for the siblings had the same mutation [Table 1].
Three children with normal renal function are on conservative measures such as hyperhydration, pyridoxine (PH 1), and crystallization inhibitors. Those in ESRD are on continuous ambulatory peritoneal dialysis, the median duration being 34.6 months.
Discussion
The estimated prevalence of PH is around 1–3 per million populations. Phenotypic heterogeneity and nonavailability of mutational analysis universally have led to its underdiagnosis. Short of mutational analysis, other investigations such as plasma and urine oxalate levels are not confirmatory.
We report a series of seven children with clinical manifestations ranging from asymptomatic nephrolithiasis to infantile oxalosis in whom genetic analysis was done. The results of the genetic analysis confirmed PH 1 in five children and PH 2 in two of them. Children with PH 2 had a milder disease phenotype. PH 3 usually being asymptomatic, was not detected in any of them. Screening of asymptomatic siblings of the index cases aided in early detection and initiation of conservative measures before the occurrence of renal dysfunction.
The PH 1 mutations c. 508G>A, c. 33dupC, and c. 731T > C have been reported as the commonest in European and African countries [67] whereas reports from North India claim c. 302T > C as the commonest.[8] The PH 2 mutations c. 103delG and c. 403_404+2delAAGT are the most common; the latter predominates in Asians.[4] However, the different set of mutations detected in our patients substantiates that these previously reported mutational hot spots may be population specific. No obvious correlation was found between the phenotype and the detection of truncating mutations. This finding is also evident from the rare kidney stone consortium PH registry. Data from the same registry also revealed that barring p.G170R, renal survival in PH 1 did not depend on the other MiR (p.G41R, p.F152I, p.I244T) mutations.[9] None of the MiR mutations were detected in our patients.
PH often presents as isolated nephrolithiasis with normal renal function. Hence, children with renal calculi should be thoroughly investigated for PH. As urinary and serum indices are unreliable in children with CKD, genetic analysis should be attempted in every child where PH is being considered clinically. The mutations being population specific, sequencing of the entire gene rather than targeted analysis will be required.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We acknowledge the efforts of the entire clinical and nonclinical staff of the Department of Pediatric Nephrology, St. John's Medical College Hospital, in identifying potential patients, procuring and transporting the blood samples. We thank Dr. Dawn Milliner and her study group, Mayo Clinic; whose research has benefitted our patients and their families to a great extent.
References
- Evidence of true genotype-phenotype correlation in primary hyperoxaluria type 1. Kidney Int. 2010;77:383-5.
- [Google Scholar]
- Primary hyperoxaluria type 2. 2008. GeneReviews. Seattle (WA): University of Washington, Seattle; Available from: http://www.ncbi.nlm.nih.gov/books/NBK2692
- [Google Scholar]
- The enzyme 4-hydroxy-2-oxoglutarate aldolase is deficient in primary hyperoxaluria type 3. Nephrol Dial Transplant. 2012;27:3191-5.
- [Google Scholar]
- Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias. Kidney Int. 2004;66:959-63.
- [Google Scholar]
- Selected AGXT gene mutations analysis provides a genetic diagnosis in 28% of Tunisian patients with primary hyperoxaluria. BMC Nephrol. 2011;12:25.
- [Google Scholar]
- Common mutation underlying primary hyperoxaluria type 1 in three Indian children. Indian J Nephrol. 2012;22:459-61.
- [Google Scholar]
- Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol. 2015;26:2559-70.
- [Google Scholar]




