

Translate this page into:
Amelogenesis Imperfecta with Distal Renal Tubular Acidosis: A Novel Syndrome?
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Amelogenesis imperfecta (AI) is a heterogeneous group of inherited dental enamel defects. It has rarely been reported in association with multiorgan syndromes and metabolic disorders. The metabolic disorders that have been reported in association with AI include hypocalciuria, impaired urinary concentrating ability, and Bartter-like syndrome. In literature, only three cases of AI and distal renal tubular acidosis (dRTA) have been described: two cases in adults and a solitary case in the pediatric age group. Here, we report a child with AI presenting with dRTA; to the best of our knowledge, our reported case is the only second such case in pediatric age group. Our case highlights the importance of recognizing the possibility of renal abnormalities in patients with AI as it will affect the long-term prognosis.
Keywords
Amelogenesis imperfecta
distal renal tubular acidosis
inherited enamel defects
nephrocalcinosis
renal tubular disorders

Introduction
Amelogenesis imperfecta (AI) is a diverse group of hereditary disorders that affect the quality and/or quantity of dental enamel. Reduced enamel thickness, typically with normal hardness, is classified as hypoplastic AI, whereas reduced hardness, discoloration with normal thickness, is termed hypermineralized AI.[1] In distal renal tubular acidosis (dRTA), kidneys are unable to acidify the urine to pH <5.5 in the presence of systemic metabolic acidosis or after acid loading as a result of impaired hydrogen ion secretion. In children, dRTA is most often a primary, hereditary condition and presents as hyperchloremic metabolic acidosis, hypokalemia, nephrocalcinosis/nephrolithiasis, polyuria, rickets, and growth retardation.[2] Early diagnosis of dRTA is critical so as to maximize growth with the treatment. Bajpai et al. have reported that maximum catch-up growth occurs if dRTA is diagnosed and treated during the first 2 years of life. Diagnosis and treatment after two years of age lead to significant height loss.[3]
AI usually occurs alone but has been rarely reported in association with multiorgan syndromes such as cone-rod dystrophy, platyspondyly, nephrocalcinosis, hypothalamo–hypophyseal insufficiency, and Kohlschutter Syndrome.[45] AI has also been rarely reported in association with metabolic disorders such as hypocalciuria, Bartter-like syndrome, and dTRA.[6789] In literature, only three cases of AI and dRTA have been described: two in adults and one in a child.[789] Here, we report a child with AI and dTRA; this is only second such case in pediatric age group.
Case Report
A 12-year-old girl was referred to the Endocrine Center at Sher-i-Kashmir Institute of Medical Sciences, Srinagar, Jammu and Kashmir, for evaluation of polyuria and short stature. From the age of 4 years, her parents had noticed abnormalities of her teeth. She is the 5th of the six siblings born to native Kashmiri consanguineous (first cousins) parents. Her younger brother had died at the age of 4 years, and he had presented with hypokalemic quadriparesis. Examination revealed prepubertal girl with severe, proportionate short stature (height 117 cm, height standard deviation [SD] score − 5.32). Her weight was 27 kg (weight SD score − 1.76). There were no clinical features of rickets. Oral examination revealed generalized pitting, yellowish-brown pigmentation of all the surfaces of the teeth, delayed exfoliation of deciduous teeth, and delayed eruption of permanent teeth [Figure 1]. Detailed ophthalmic examination and pure-tone audiometry were normal. Radiographically, the radiodense enamel was absent in all the teeth with normal radiodensity of dentin, large, and open pulp canals. Few retained deciduous teeth and impacted canines and premolars were seen. The clinical and radiographic features led to the diagnosis of hypoplastic AI.
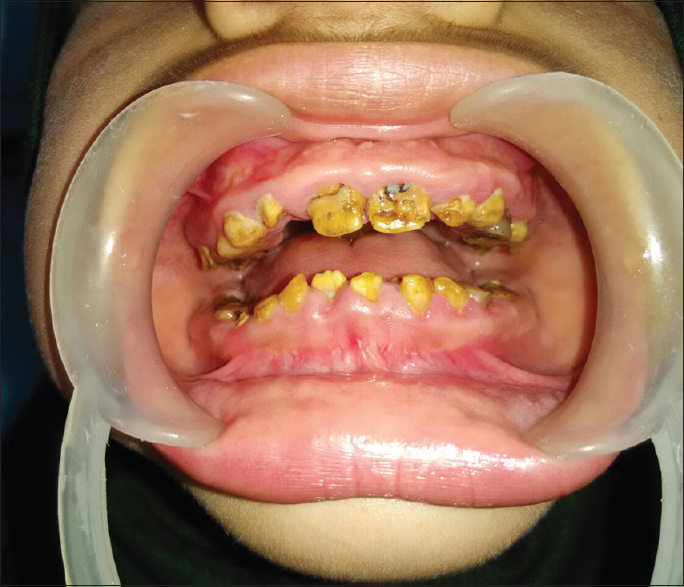
- Photograph of the patient showing generalized pitting, yellowish-brown pigmentation, and reduction of enamel thickness
Investigations revealed a normal anion gap metabolic acidosis (serial serum pH 7.19, 7.20, and 7.22 and anion gap 11, 14, and 15), alkaline urine (serial urine pH 6.7, 6.5, and 7), and hypokalemia (serial serum K+ 1.85, 1.52, and 2.8 mEq/L). Urine anion gap was positive. Serum osmolality was 277 and urine osmolality was 128 mosm/kg. Routine lab investigations including blood glucose, serum calcium, serum phosphorus, alkaline phosphatase, thyroid function test, renal, and liver function tests were normal. The urinary calcium excretion was increased (236 mg/day). Urine analysis did not reveal glycosuria, proteinuria, or aminoaciduria indicating normal proximal tubular reabsorption. Serum 25-OH D levels were normal (37 ng/ml).
Bilateral nephrocalcinosis was identified by means of ultrasonography [Figure 2]. Radiograph of left wrist did not show any evidence of rickets, and bone age was normal (according to Greulich and Pyle Atlas).

- Renal ultrasonography showing bilateral nephrocalcinosis (white arrows indicating nephrocalcinosis)
On the basis of persistent normal anion gap, metabolic acidosis with alkaline urine, hypokalemia, hypercalciuria, and bilateral nephrocalcinosis, a diagnosis of dRTA was made. The diagnosis was confirmed by ammonium chloride test, and the serial urine pH after oral intake of ammonium chloride was 7.3, 7.5, and 6.9. We further investigated her for secondary causes of RTA, and autoimmune profile including antinuclear antibody was negative. She was started on oral potassium citrate supplementation. Treatment with potassium citrate resulted in correction of hypokalemia, systemic acidosis, and marked relief of polyuria.
Discussion
The AI can be transmitted by an autosomal dominant, autosomal recessive, or X-linked mode of inheritance. Mutations in five genes (AMELX, ENAM, KLK4, DLX3, and MMP20) have been reported to cause AI.[10] In children, dRTA is most often a primary inherited condition and can be essentially of three types: autosomal dominant dRTA (the commonest form) and autosomal recessive dRTA with and without sensorineural deafness.[2] In autosomal recessive form, mutations involve subunits of the H+-ATPase proton pump while autosomal dominant form results from mutations involving the chloride-bicarbonate exchanger.[2] Congenital hypothyroidism has also been associated with dRTA in children.[11] Metabolic acidosis due to RTA can be differentiated from gastrointestinal loses on the basis of urine anion gap. In RTA, urine anion gap is positive while in gastrointestinal causes of acidosis it is negative.
AI has rarely been associated with renal parenchymal and renal tubular disorders such as bilateral nephrocalcinosis, hypocalciuria, impairment of renal concentration, hypokalemic metabolic alkalosis (Bartter-like syndrome), and impairment of urinary tubular acidification. The syndrome of AI with nephrocalcinosis is known as enamel-renal syndrome.[5] This is a rare syndrome, and some of these cases can progress to renal insufficiency.[12] The association of AI with RTA is exceptionally rare, and till date, only two cases in adults and a solitary case in the pediatric age group have been reported.[789] Most of these cases including our case have been reported in consanguineous families, suggesting an autosomal recessive mode of inheritance.
How can we link amelogenesis imperfecta with renal tubular acidosis?
Different renal proteins previously known to play a role in the systemic pH homeostasis in mammals have also been described to be expressed during amelogenesis: the carbonic anhydrase II, acid–base exchangers AE2, NBCe1, and NHE1.[1314] H+-ATPase proton pump is expressed at high density on the luminal membrane of α intercalated cells of cortical collecting tubule. H+-ATPase proton pump is also expressed in ameloblasts at maturation.[13] Mutation of H+-ATPase proton pump is well known to cause autosomal recessive form of dRTA.[15] There is a strong possibility that mutation of H+-ATPase proton pump can cause both AI and dRTA. On clinical grounds, there is a possibility that our patient may be harboring this mutation because only this mutation can explain the association of AI with dRTA. Due to lack of facilities, we have not tested this mutation in our patient.
Another hypothesis that may link AI with RTA is that many of the dental proteins that were thought to be tissue-specific may also be expressed in nondental tissues such as renal tissues. For example, DLX3, a homeobox protein, is also expressed in the kidney. Further research concerning these proteins is necessary to determine whether they play a pivotal role in renal tubular disorders.
The orodental phenotype of AI is often complex and difficult to manage. Gingival hyperplasia is managed by gingivectomy. Erupted teeth can be restored with conventional treatments such as ceramic crowns. Severe cases characterized by a large number of nonerupted teeth need complete rehabilitation with overdentures.
Conclusion
Our report concerns a case of the rare syndrome of AI and dRTA and, hence, highlights the importance of detailed renal evaluation in patients with AI. An early diagnosis provided by the pathognomonic oral phenotype can lead to a better long-term renal prognosis. Awareness of the possibility of such an association is very essential among dentists to ensure an early referral of these patients to physicians. To conclude, the present case highlights the importance of a thorough medical history, systemic examination, and detailed renal function tests including assessment of renal tubular functions in all patients with AI.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Amelogenesis imperfecta: A classification and catalogue for the 21st century. Oral Dis. 2003;9:19-23.
- [Google Scholar]
- Long-term outcome in children with primary distal renal tubular acidosis. Indian Pediatr. 2005;42:321-8.
- [Google Scholar]
- Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am J Hum Genet. 2009;84:266-73.
- [Google Scholar]
- The relationship of amelogenesis imperfecta and nephrocalcinosis syndrome. Med Oral Patol Oral Cir Bucal. 2009;14:e579-82.
- [Google Scholar]
- Enamel-renal syndrome associated with hypokalaemic metabolic alkalosis and impaired renal concentration: A novel syndrome? Nephrol Dial Transplant. 2006;21:2959-62.
- [Google Scholar]
- Amelogenesis imperfecta and distal renal tubular acidosis presenting as hypokalemic periodic paralysis. J Assoc Physicians India. 1999;47:1205-6.
- [Google Scholar]
- Amelogenesis imperfecta with renal disease – A report of two cases. J Oral Pathol Med. 2007;36:625-8.
- [Google Scholar]
- Distal renal tubular acidosis and amelogenesis imperfecta: A rare association. Indian J Nephrol. 2013;23:452-5.
- [Google Scholar]
- Genes and related proteins involved in amelogenesis imperfecta. J Dent Res. 2005;84:1117-26.
- [Google Scholar]
- Congenital hypothyroidism due to thyroid dysgenesis associated with distal renal tubular acidosis and hypoplastic kidney, an unknown association. Thyroid Res Pract. 2016;13:40-2.
- [Google Scholar]
- Localization of H(+)-ATPase and carbonic anhydrase II in ameloblasts at maturation. Calcif Tissue Int. 1994;55:38-45.
- [Google Scholar]
- Ion transporters in secretory and cyclically modulating ameloblasts: A new hypothesis for cellular control of preeruptive enamel maturation. Am J Physiol Cell Physiol. 2010;299:C1299-307.
- [Google Scholar]
- Molecular cloning and characterization of novel tissue-specific isoforms of the human vacuolar H(+)-ATPase C, G and d subunits, and their evaluation in autosomal recessive distal renal tubular acidosis. Gene. 2002;297:169-77.
- [Google Scholar]




