

Translate this page into:
Novel Heterozygous Mutations of Congenital Thrombotic Thrombocytopenic Purpura: A Rare Case Report
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Hereditary thrombotic thrombocytopenic purpura (TTP) is a genetic condition caused by mutations in ADAMTS13 gene, leading to very low levels of ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type I domain 13) activity. It is a rare condition associated with multiple reported mutations. Here, we describe a case of hereditary TTP with a compound novel heterozygous mutation along with secondary focal segmental glomerulosclerosis. The patient responded clinically to plasma infusions with resolution of thrombocytopenia, stabilization of renal function, and control of blood pressures. Genetic analysis of the entire family helped in the characterization of the inheritance of this mutation. Our case illustrates the need for focused genetic analysis in a subset of patients presenting with features of TTP to decide the therapeutic plan and manage accordingly.
Keywords
Novel mutation
plasma infusion
secondary focal segmental glomerulo sclerosis
thrombotic thrombocytopenic purpura

Introduction
Thrombotic thrombocytopenic purpura (TTP) is classically characterized by thrombotic microangiopathy, renal failure, neurological dysfunction, and thrombocytopenia. TTP is now a disease with a well-understood pathophysiology, characterized by platelet aggregation due to ultra large von Willebrand factor (vWF) multimers, which occur due to ADAMTS13 deficiency.[1] It can be hereditary or acquired. Hereditary/congenital TTP or Upshaw–Shulman syndrome (USS, online Mendelian Inheritance in Man reference 274150) is caused by mutations in ADAMTS13 gene leading to severe deficiency of ADAMTS13 activity. The deficiency of ADAMTS13 activity can also be caused by inhibitory antibodies targeting ADAMTS13, which is the basis of acquired TTP. Here, we describe a case of hereditary TTP with compound heterozygous mutations in ADAMTS13 gene, which presented in the second decade of life.
Case Report
A 21-year-old male with no prior comorbidities was first symptomatic in January 2016 with complaints of intermittent, low-grade fever and vomiting of 1-month duration. Patient was the youngest of five siblings, and was born of a nonconsanguineous marriage. He was evaluated in a local hospital and was found to be normotensive with a platelet count of 49000/cu.mm, hemoglobin of 11.0 g/dl, and total leucocyte count of 6100/cu.mm. He underwent bone marrow biopsy for the same, which showed increased number of megakaryocytes. The patient was started on oral steroids which he took irregularly; he also received platelet transfusions over 1 month because of persistent low platelet counts. Renal function tests at initial evaluation revealed blood urea of 52 mg/dl and serum creatinine of 1.2 mg/dl. After 2 months of initial presentation, the patient was admitted with accelerated hypertension, anemia (hemoglobin, 9.0 gm/dl), and thrombocytopenia (platelet count, 22000/cu.mm). On evaluation, there were >1% schistocytes in peripheral smear and renal dysfunction, with serum creatinine increased to 2.8 mg/dl. Urine examination did not show any proteinuria or active sediments. Serum bilirubin was 1.3 mg/dl, and lactate dehydrogenase level was 593 U/L. Ultrasound kidneys and renal doppler were normal. There were no abnormalities detected in coagulation profile. The patient was suspected to have TTP and was started on plasma exchange with fresh frozen plasma. His renal function improved, and serum creatinine decreased to 1.5 mg/dl over the next 1 month. Renal biopsy was deferred on account of thrombocytopenia. ADAMTS13 activity was less than 3% (reference range, 68–163%), and ADAMTS13 inhibitor assay was negative (<0.4 Bethesda titre). The patient was diagnosed as hereditary TTP, and was discharged with advice to continue fresh frozen plasma infusion every 3 weeks. The fundus examination revealed exudative retinopathy secondary to hypertension. On follow-up, his serum creatinine remained stable at 1.4–1.5 mg/dl for over 1 year and increased to 2.2 mg/dl in February 2018. His routine urine examination showed 2+ protein and 0–2 red blood cells per high power field. Urine spot protein creatinine ratio was 1.1, and his complete blood picture was normal (hemoglobin, 14.1 g/dl, total leucocyte count, 6700/cu.mm, and platelet count 346000/cu.mm). On light microscopy, renal biopsy comprised single linear core of cortex with 20 glomeruli, five of which were globally sclerosed, and three showed segmental sclerosis with one showing overlying podocytic hypertrophy. The remaining glomeruli showed uniform glomerulomegaly and mild mesangial proliferation. There was no proliferative or exudative activity, crescents, necrotizing lesion, or capillary wall thickening. Interstitial fibrosis and tubular atrophy amounted to 20–25% of the biopsied cortex. Artery showed mild fibrointimal and medial thickening. Immunofluorescence showed nonspecific deposition of IgA (2+), C3 (1-2+), kappa (1+), and lambda (1+) in mesangium, whereas it was negative for IgG, IgM, and C1q. Electron microscopy revealed focal effacement of foot process (approximately 10%), with normal thickness of glomerular basement membrane and no electron dense deposits. Hence, overall features were consistent with secondary focal segmental glomerulosclerosis, with mild tubulointerstitial chronicity. Although immunofluorescence revealed 2+ IgA staining, on account of normal mesangial cells with absence of electron dense deposits in electron microscopy, it was considered to be a nonspecific finding. In view of normal ADAMTS13 inhibitor assay and low ADAMTS13 activity, mutation analysis was done which revealed two heterozygous mutations in ADAMTS13 gene [exon 10, c.1201G>A (p. Gly401Arg) and exon 25 c.3265G>T (p. Gly1089Trp)], which was suggestive of familial TTP. Pedigree chart of our patient is shown in Figure 1. Mutation analysis of parents revealed heterozygous mutations in both. The mother had the same mutation at exon 10 whereas the father had the same mutation at exon 25 [Table 1]. The patient is on regular fresh frozen plasma transfusions with blood pressure well controlled on antihypertensive drugs and serum creatinine stable at 2.2 mg/dl.
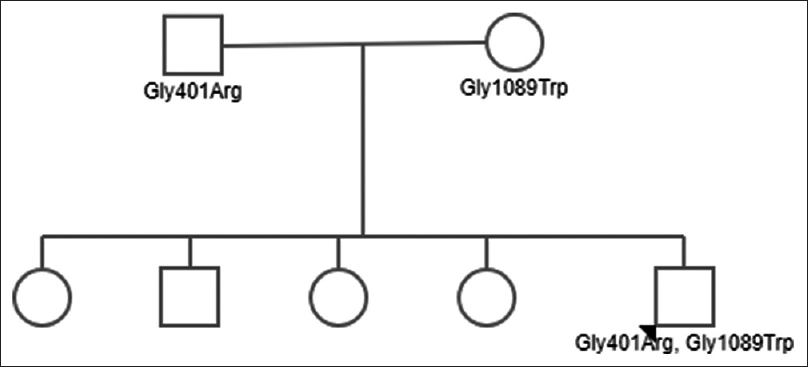
- Pedigree chart of the index patient with genetic mutations
| Subject | Gene | Location of mutation | Variant | Detection | Clinical condition |
|---|---|---|---|---|---|
| Mother | ADAMTS13 | Exon 10 | c.1201G>A (p.Gly401Arg) | Absent | Asymptomatic |
| Exon 25 | c.3265G>T (p.Gly1089Trp) | Present | |||
| Father | ADAMTS13 | Exon 10 | c.1201G>A (p.Gly401Arg) | Present | Asymptomatic |
| Exon 25 | c.3265G>T (p.Gly1089Trp) | Absent | |||
| Index case | ADAMTS13 | Exon 10 | c.1201G>A (p.Gly401Arg) | Present | Symptomatic |
| Exon 25 | c.3265G>T (p.Gly1089Trp) | Present |
Discussion
TTP was first described by Eli Moschcowitz, who reported the case of a 16-year-old girl who died after acute-onset hemolytic anemia, thrombocytopenia, petechiae, fever, and severe neurological symptoms.[2] The basic mechanism of TTP is low ADMTS13 activity, because of genetic defects causing low enzyme activity or acquired inhibitors of ADMTS13. Congenital TTP is a rare genetic condition with an estimated incidence of 4–10 cases per million/year.[3] Congenital TTP is characterized by low ADAMTS13 activity in the absence of antiADAMTS13 autoantibodies. ADAMTS13 is a metalloprotease that specifically cleaves vWF multimers. vWF is essential for platelet adhesion and aggregation under high shear stress conditions. ADAMTS13 is mainly synthesized by the liver, and is also expressed on the podocytes of glomeruli. In glomerular capillaries, the proteolytic activity could have a protective effect preventing deposition of platelets along capillary lumina under conditions of high shear stress present in glomeruli.[4] ADAMTS13 is essential to modify the size of vWF multimers. This metalloprotease is encoded by a gene on chromosome 9q34.[5] Mutation in this gene is critical in the pathogenesis of hereditary TTP. More than 130 ADAMTS13 gene mutations have been reported worldwide.[36789] ADAMTS13 consists of an N-terminal signal peptide metalloproteinase, a disintegrin-like and thrombospondin-type 1 (TSP1) motif, a cysteine rich/spacer domain and additional TSP motifs, and CUB (complement C1r/C1s, Uegf, Bmp1) domains. Patients with ADAMTS13 gene defects present from birth, but some patients manifest in adulthood.[710] This suggests the need of further genetic and environmental factors for the disease to manifest. Environmental triggers may include infection, pregnancy, surgery, bone marrow transplant, and heavy alcohol intake.[3] Our patient presented in the second decade of life with a febrile illness at the disease onset. He developed the classical features of TTP with hemolytic anaemia and mild renal dysfunction. Although renal dysfunction improved with plasma exchange therapy, it did not reach the normal baseline in this patient. He required regular infusions of fresh frozen plasma consistent with severe deficiency of ADAMTS13. Stopping the plasma infusions led to a drop in platelet count and uncontrolled blood pressure on the same drugs with which his blood pressures were controlled previously. Plasma infusion (10–15 ml/kg) or factor VIII concentrate can be used to normalize ADAMTS13 levels in these patients.[11] Mutation analysis revealed that the patient carried a compound heterozygous mutation of ADAMTS13 gene with a missense variation in exon 10, which was inherited from his mother that results in amino acid substitution of arginine for glycine at codon 401. There was another heterozygous missense variation in exon 25 of the ADAMTS13 gene, which was inherited from his father that results in the amino acid substitution of tryptophan for glycine at codon 1089. Both the variations were in the TSP1 domain of ADAMTS13 protein. Most missense mutations that have been investigated appear to cause ADAMTS13 deficiency by impairing protein secretion.[3] Renal biopsy showed features of secondary Focal Segmental Glomerulo Sclerosis (FSGS), which may be secondary to hypertension, as there was focal effacement of foot process (approximately 10%) on electron microscopy. Our case is interesting from a clinical point of view because, despite having a congenital condition, presentation was seen in adulthood possibly due to a trigger. Renal biopsy was not done at the initial presentation in view of clinical response and presence of thrombocytopenia, which possibly could have revealed features of thrombotic microangiopathy.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Hyaline thrombosis of the terminal arterioles and capillaries: A hitherto undescribed disease. Proc N Pathol Soc. 1924;24:21-4.
- [Google Scholar]
- Inherited ADAMTS13 deficiency (Upshaw-Schulman syndrome): A short review. Thromb Res. 2014;134:1171-5.
- [Google Scholar]
- Phenotypic expression of ADAMTS13 in glomerular endothelial cells. PLoS One. 2011;6:e21587.
- [Google Scholar]
- Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488-94.
- [Google Scholar]
- ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura. Hum Mutat. 2010;31:11-9.
- [Google Scholar]
- Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost. 2011;9(Suppl 1):283-301.
- [Google Scholar]
- Ten candidate ADAMTS13 mutations in six French families with congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome) J Thromb Haemost JTH. 2004;2:424-9.
- [Google Scholar]
- Pathophysiology of thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. J Thromb Haemost. 2018;16:618-29.
- [Google Scholar]
- A case of congenital TTP presenting with microganiopathy in adulthood. BMC Hematol. 2014;14:16.
- [Google Scholar]
- How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br J Haematol. 2014;164:759-66.
- [Google Scholar]




