

Translate this page into:
Role of Genetic Screening in the Management of Familial Focal Segmental Glomerulosclerosis: A Tale of Two Sisters
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Sir,
We report the genetic screening results in two siblings with steroid-resistant nephrotic syndrome (SRNS) performed at different time points in the clinical course, which provided a unique opportunity to highlight the utility of genetic diagnosis at the right time. A subset of children with SRNS has an underlying mutation in the podocyte-specific genes. Recent studies analyzing genetic mutations in children with SRNS found 10–32% of the SRNS cohort, harboring a disease-causing variant with a higher proportion in familial cases (67%) as compared with sporadic cases (25%).[12] Genetic testing in these patients will not only aid in diagnosis and prognostication of the disease but also help in limiting the exposure to expensive immunosuppression therapies and related complications.
We herein present a consanguineous Indian family with two affected children suffering from SRNS. The elder sibling (P1) presented with SRNS at 3 years of age and was diagnosed to have FSGS (not otherwise specified) on renal biopsy. She initially received empirical immunosuppression therapy with steroids, cyclosporine, mycophenolate mofetil, and antiproteinuric drugs over 30 months with no response in proteinuria. She also required multiple hospitalizations for infections and for hypertensive encephalopathy with seizures because of posterior reversible encephalopathy syndrome. She progressed to end stage renal disease (ESRD) by 6 years of age requiring dialysis for 12 months along with a unilateral nephrectomy for uncontrolled hypertension and proteinuria. The child succumbed to severe sepsis at the age of 7 years. The genetic screening for NPHS2 and WT1 using Sanger sequencing was performed when the child was in ESRD. A homozygous pathogenic variant c.211C>T; p.R71X in exon 1 of the NPHS2 gene was identified. Within a year, the younger sibling was born and was kept under close monitoring for proteinuria. Younger sibling (P2) was diagnosed as a case of nephrotic syndrome at the age of 3.5 years on routine urine screening, although she had no edema and renal biopsy-revealed FSGS. Sanger sequencing revealed the same homozygous pathogenic variant c.211C>T; p.R71X in exon 1 of the NPHS2 gene, which was identified in the older sibling. In view of genetic diagnosis and family history, we presumed a poor response to steroid therapy and hence initiated on tacrolimus as a first-line therapy. She received tacrolimus for 18 months without reduction in proteinuria. In view of the genetic diagnosis, prolonged course of tacrolimus was not considered. The child progressed to CKD stage 3 by 7 years of age. The child did not have any hospitalization for infections or medication-related adverse effects. Parents of the siblings were identified to be heterozygous carriers for c.211C>T; p.R71X variant, compatible with recessive inheritance [Figure 1]. In addition, the mother who was the potential donor did not harbor the R229Q variant, which increases the risk of proteinuria in adulthood.
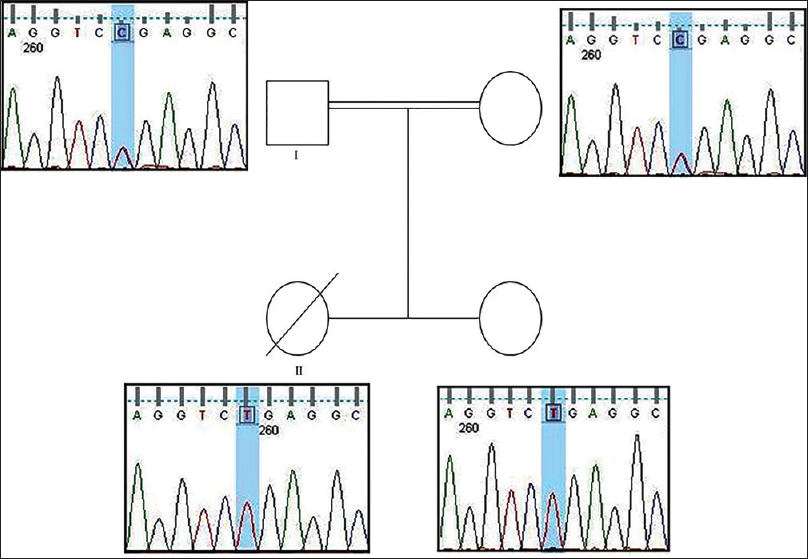
- Pedigree of a family with aNPHS2 mutation. Chromatogram of DNA sequence of NPHS2 exon 1 – c.211C>T; p.R71X. (I) Parents were identified to be heterozygous carriers for c.211C>T; p.R71X variant. (II) The siblings were identified to have the homozygous missense mutation c.211C>T, resulting in a termination codon p.R71X
The timing of genetic screening in the siblings reveals the utility of genetic screening. Besides assisting in molecular diagnosis, genetic testing also helped in deciding the treatment and in prognostication. Genetic diagnosis in the elder sibling helped us plan timely screening for proteinuria in the younger child. The genetic testing at the time of diagnosis helped to individualize immunosuppressive treatment. With the identification of the pathogenic variant in the sibling 2 at time of diagnosis of nephrotic syndrome steroid was not used for the treatment; instead, tacrolimus was given as the first-line treatment, thus avoiding the side effects of steroids. Tacrolimus was considered in this child as there were reports of partial response to calcineurin inhibitors in those with a genetic cause of SRNS.[3] The genetic information helped in predicting the response to tacrolimus and counseling the parents at the time of initiation of treatment about the efficacy of treatment. As expected, there was no significant response to tacrolimus. Further immunosuppression was avoided, which helped in reducing the morbidity because of serious infectious complications and adverse effects of medications. Genetic testing in P2 at the time of diagnosis was also a helpful factor in predicting more accurate prognosis and effective counseling as compared to P1. P2 is now being worked up for renal transplant. The information from genetic testing not only helped in assessing the risk of recurrence in the patient but also facilitated donor evaluation. Although the mother who was also the donor was a carrier of the same variant, she did not harbor the R229Q variant, which increases the risk of proteinuria in adulthood.
The detection of mutations in children with SRNS is not only of clinical benefit for the child but also in choosing a potential donor who will not have the risk of developing proteinuria later in life.[4]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patients have given their consent for their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
This study is financially supported by Indian Council of Medical Research (3/1/2/6-RCH; IRIS ID No. 2012-26950).
Conflicts of interest
There are no conflicts of interest.
References
- Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. J Am Soc Nephrol. 2015;26:230-6.
- [Google Scholar]
- Podocyte-associated gene mutation screening in a heterogeneous cohort of patients with sporadic focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2014;29:2062-9.
- [Google Scholar]
- Non-immunologic mechanisms of calcineurin inhibitors explain its antiproteinuric effects in genetic glomerulopathies. Pediatr Nephrol. 2010;25:1197-9.
- [Google Scholar]
- Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017;94:884-90.
- [Google Scholar]




