

Translate this page into:
Burning Feet, Dilated Heart and Failed Kidneys
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Fabry's disease, X-linked lysosomal storage disease, results from deficient activity of alpha galactosidaseA (α-GalA). Renal manifestation usually begins at third decade of life. We report a 16 year male with initial presentation with end stage renal disease (ESRD) and the diagnosis confirmed by presence of myeloid bodies on electron microscopy of kidney biopsy and low serum α-GalA level.
Keywords
Alpha galactosidase
Fabrys disease
myeloid bodies

Introduction
Fabry's disease overall accounts for about 0.02% of all causes of ESRD.[1] Vague symptomatology and a lack of specific physical findings often lead to delay in the diagnosis of this rare disease. ESRD is seen usually in fourth to fifth decade of life.[2] We report a 16 year old adolescent male with an aim to highlight the uncommon presentation of Fabry with ESRD in adolescence and the utility of renal biopsy in the diagnosis of Fabry's disease.
Case Report
A 16-year-old male, presented with five days history of progressively worsening dry cough with exertional dyspnoea, three days after the onset of these symptoms he developed edema and oliguira. There was no history of hematuria, headache, seizures, rashes, joint pain, hearing or visual abnormalities. He did not give any history of repeated urinary tract infection, and had a good scholastic performance. There was no family history of any renal disease. Past medical history was uneventful except for burning sensation in hand and feet on and off for past two years. Clinically he was under nourished, weight: 39 kgs (<3rd centile); Height: 160 cms (3rd centile). On examination there was mild pallor, facial puffiness, ascites and limb edema. He had stage II hypertension, with raised JVP, tender hepatomegaly, basal crepitation, tachycardia and gallop cardiac rhythm. Fundus examination was normal. There was no renal bruit. Neurolog ical and musculoskeletal examination was normal.
Investigations showed hemoglobin of 9.1 gm/dl and total leucocyte count of 14200/cu.mm with normal differentials and platelet count. Complete urine examination revealed 2+ protein with no RBC/WBC or casts. Along with elevated blood urea (303 mg/dl) and serum creatinine (9.3 mg/dl), there was hyponatremia (129 mmol/L) and hyperkalemia (6.0 mEq/L). The liver enzyme were also abnormal with raised AST (265U/l) and ALT (686U/l). Renal ultrasound showed right kidney (8 × 4.4) cms and left kidney (9.6 × 5.2) cms with both showing increased cortical echogenicity and loss of cortico-medullary differentiation.
Bone chemistry showed Calcium, Phosphorus and ALP of 5.1 mg/dl, 8.9 mg/dl and 170 IU/L respectively. Parathormone (30.55 ng/ml) and 25-OH D3 level (68.43 ng/ml) were normal. Echocardiogram demonstrated dilated cardiomyopathy with, global left ventricle hypokinesia and pulmonary arterial hypertension. Work up for autoimmune and vasculitis (C3, C4, ANCA and ANA) was normal. Micturating cystourethorgram done in view of discrepancy in renal sizes ruled out reflux. Following the detailed clinical evaluation and investigations, a possibility of chronic kidney disease (CKD) stage-V probably due to nephronophthisis or chronic glomerulopathy or chronic interstitial nephritis with uremic neuropathy or vitamin B 12 deficiency (421 pg/ml) were considered.
In view of ESRD with fluid overload, uraemia and hyperkalemia, regular hemodialysis along with supportive measures were initiated. Hypertension was controlled on four anti-hypertensive medications. He also required oral digoxin and sildenafil in renal corrected doses in view of poor cardiac status and severe pulmonary hypertension. However, he continued to have episodes of severe burning and pain sensation over both lower limbs not relieved with adequate hemodialysis, analgesics or vitamin B12 supplementation. A possibility of Fabry's disease was kept in view of cardiac involvement, renal failure and acroparasthesias. Electroneuromyography was suggestive of bilateral common peroneal motor and sensory axonal neuropathy in the lower limbs and bilateral sural sensorial axonal neuropathy.
A renal biopsy was performed which showed visceral epithelial proliferation in two of the viable glomeruli with prominent cytoplasmic vacuolization in all the visceral epithelial cells. There was extensive interstitial fibrosis and tubular atrophy, involving more than 75% of the core. Marked fibrous intimal proliferations of arteries wereseen. The electron microscopy revealed podocytes stuffed with myeloid bodies [Figure 1]. Hence a possibility of Fabrys disease was considered and was confirmed by absence ofalpha galactosidase activity in blood. Genetic testing for a molecular diagnosis was not performed in view of cost constraints. In addition, Enzyme replacement therapy (ERT) was not started as parents could not afford the therapy. The child continues to be on thrice weekly hemodialysis.
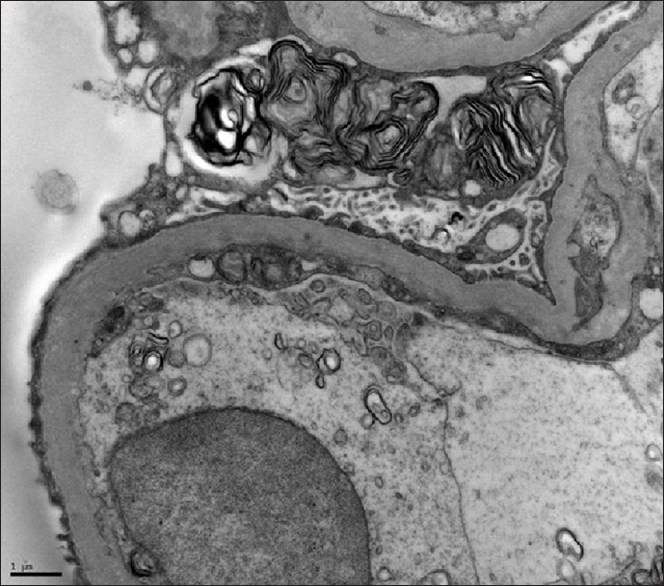
- Electron microscopy of kidney showing myeloid bodies
Discussion
Fabry disease is an X-linked inheritedlysosomal storage disorder due to mutations in α-Gal A gene mapped on Xq22.1. The disease has worldwide prevalence of 1 in 100,000 to 500,000 live births. The diagnosis of Fabry's disease among children is difficult in the absence of a family history and non-specific clinical manifestation which includes acroparasthesias, hypohidrosis, angiokeratomas, corneal dystrophy, gastrointenstinal distress, cardiomyopathy, renal failure and cerebrovascular events. The neuralgic pain is often misdiagnosed as gout or other rheumatological disorder. Classical angiokeratomas may be absent as seen in our index case. Fabry disease accounts for about 0.2-1.2% of patients with ESRD on hemodialysis, 4.9% of men and 2.4% of women with cryptogenic stroke and 6.3% of late onset hypertrophic cardiomyopathy.[345]
Renal impairment begins with microalbuminuria and proteinuria in the third decade of life with gradual development of azotemia, and ESRD by fourth to fifthdecade.[3] Interestingly our index child presented with ESRD as the initial presentation. Most of the cases that has been described from the country had presented with non-renal manifestations. Gayathri et al. reported a 19 yr old boy who presented with acroparasthesias, angiokeratomas and corneal opacities and nerve biopsy by electron microscopy revealed lamellated inclusions in the smooth muscles, perineurial and endothelial cells characteristic of Fabrys disease.[6] This child however did not have cardiac and renal involvement as our child.
Surjushe et al. reported a 12 yr old male child with Fabrys disease with marfanoid features with renal involvement in the form of proteinuria and hypertension.[7] Phadke et al. also reported a 13 year old male with Fabry's disease presenting with acroparasthesias as a predominant manifestation. Familial screening showed his younger brother had Fabry'swith gastro intestinal symptoms and mother having corneal deposits and proteinuria.[8]
In the absence of a genetic diagnosis, a diagnosis could be potentially made on renal biopsy. It is uncommon to perform a renal biopsy in child with ESRD due to higher risk bleeding complications and low diagnostic yield. Since, the kidney sizes were normal in this child and in absence of a clear diagnosis, a renal biopsy was performed. On light microscopy glomerular visceral epithelial cells are enlarged and vacuolated, and tubules and blood vessels are also abnormal. Immunofluorescence is usually negative, however in advance stages there may be IgM, C3 and C1q deposits in capillary wall and mesangial regions. In electron microscopy, cellular Gb3 inclusion (myelin figures) within lysosomes surrounded by a single membrane can be found in all cells mainly podocytes and endothelial cells.[3]
Early diagnosis of Fabry much before the onset of severe renal failure is important as two enzyme preparations: Fabrazyme and Replagal, using recombinant human a-Gal A are available for treatment. The clinical trials, using these enzymes have shown to stabilize the renal function for those with GFR of greater than or equal to 60 ml/min per 1.73 m2 and also results in relatively slow decline in renal function below this GFR over a period of about 4.5 yrs.[910] However among those who are diagnosed late and have already progressed to ESRD, renal transplant is the first choice as it does not recur in the allograft.
In conclusion clinician should suspect disease in children presenting with chronic kidney disease who also have dilated cardiomyopathy and acroparasthesias that persists despite adequate dialysis and correction of vitamin B12 deficiency. Renal biopsy with appropriate precautions can help in the diagnosis of Fabry's disease.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
Dr. Gurinder Kumar, Fellow in Pediatric Nephrology, Bengaluru, for participation in the management of the case.
References
- Patients with Fabry disease on dialysis in the united states. Kidney Int. 2002;61:249-55.
- [Google Scholar]
- Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105:1407-11.
- [Google Scholar]
- Prevalence of Fabry disease in patients with cryptogenic stroke: A prospective study. Lancet. 2005;366:1794-6.
- [Google Scholar]
- Fabry's disease: An ultrastructural study of nerve biopsy. Ann Indian Acad Neurol. 2008;11:182-4.
- [Google Scholar]
- Anderson-Fabry's disease with marfanoid features. Indian J Dermatol Venereol Leprol. 2008;74:389-91.
- [Google Scholar]
- Fabry disease: A treatable lysosomal storage disorder. Natl Med J India. 2009;22:20-2.
- [Google Scholar]
- Enzyme replacement therapy in Fabry disease: A randomized controlled trial. JAMA. 2001;285:2743-9.
- [Google Scholar]
- Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease. N Engl J Med. 2001;345:9-16.
- [Google Scholar]




