

Translate this page into:
Uncommon Presentation of Atypical Hemolytic Uremic Syndrome: A Case Report
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Atypical hemolytic uremic syndrome (aHUS) is an ultra-rare disease characterized by microangiopathic hemolytic anemia, thrombocytopenia and renal damage. Its presentation as nephrotic syndrome (NS) during first year of life is uncommon; we describe a child with clinical and laboratory findings of NS whose renal biopsy revealed thrombotic microangiopathy (TMA). A previously healthy 4-month-old male was admitted with severe dehydration, diarrhea and anuria. Laboratory results showed electrolyte disturbances, increased serum creatinine, anemia without schistocytes, thrombocytosis, normal lactic dehydrogenase (LDH) levels, hypoalbuminemia hypercholesterolemia and decreased C3 levels. After rehydration hematuria and massive proteinuria were also documented and an initial diagnosis of NS of the first year was established. Studies seeking for infectious agents were negative. During hospitalization he continued to be oligo-anuric needing dialysis and a renal biopsy was performed, which showed TMA findings. We here considered the diagnosis of aHUS and started plasma infusions as a bridge until starting eculizumab. After two infusions urine output improved leading to discontinuation dialysis. The diagnoses of STEC infection and thrombocytopenic thrombotic purpura were ruled out. Factor B, H, I and properdin levels were normal. Antibodies against CFH negative were negative. Screening for genes causative of aHUS detected a heterozygous variant in CFHR3 of uncertain significance. On day 20, treatment was switched to eculizumab, which induced a progressive remission of the NS. This case outlines the need for a heightened diagnosis suspicion of this already rare disease since early initiation of eculizumab therapy improves its prognosis.
Keywords
aHUS
eculizumab
nephrotic syndrome
thrombotic microangiopathy

Introduction
Atypical hemolytic uremic syndrome (aHUS) is an ultra-rare disease characterized by microangiopathic hemolytic anemia, thrombocytopenia and renal damage due to a systemic microangiopathic process in most patients,[12] but ≈25% of them do not have this classic triad on presentation.[3] In at least 50% of patients, aHUS is caused by genetic or acquired (autoantibodies) deregulation and uncontrolled activation of the alternative pathway of the complement system leading to endothelial damage. Clinical onset is often abrupt, although in 20% of patients may be progressive accompanied by subclinical anemia, fluctuating thrombocytopenia and preserved renal function.[1] Presentation as nephrotic syndrome during first year of life is uncommon.[1] We describe here a child with edema, ascites, hypoalbuminemia and serum lipid alterations without hemolytic findings on peripheral smear whose renal biopsy revealed thrombotic microangiopathy (TMA).
Case Report
A previously healthy 4-month-old male born at 38 weeks of gestation with adequate birth weight was admitted to our hospital with diarrhea and anuria for the last 12 hrs. He was the first child of non-consanguineous marriage. Three weeks before admission he had received rotavirus, pneumococcal conjugate, pentavalent and inactivated polio vaccines. Before admission he developed fever and watery diarrhea that were treated symptomatically. Upon arrival, he was pale, somnolent and severely dehydrated; thus, intravenous saline solution was immediately infused. Initial laboratory results showed pH 7.32, bicarbonate 13 mEq/L, sodium 107 mEq/L, potassium 6.5 mEq/L and chloride 86 mEq/L with a glomerular filtration rate estimated by Schwartz formula (eGFR) of 55 ml/min/1.73 m2. He also had anemia without hemolytic forms on peripheral smear and thrombocytosis, normal lactic dehydrogenase (LDH) levels, hypoalbuminemia and hypercholesterolemia [Table 1]. During rehydration he suffered a generalized tonic-clonic seizure, which was attributed to severe hyponatremia. A magnetic resonance of the brain was normal. After 24 hrs the patient had generalized edema, blood pressure 114/70 mmHg and oligoanuria. Subsequent laboratory evaluations revealed improved renal function, microcytic hypochromic anemia without schistocytes and elevated platelets count. C3 level were decreased and C4 values were within the normal range. Urinalysis showed hematuria and proteinuria in nephrotic range [Table 1]. Abdominal ultrasound demonstrated enlarged echogenic kidneys and ascites.
| Parameters | Normal value | Admission | Day 1 | Day 6 | Day 14 | Day 39 | Day 46 | Day 65 | Day 270 |
|---|---|---|---|---|---|---|---|---|---|
| Urea (mg/dL) | 18-40 | 96 | 87 | 90 | 60 | 14 | 8 | 10 | 20 |
| Creatinine (mg/dL) | 0.17-0.42 | 0.52 | 0.28 | 1.17 | 0.47 | 0.17 | <0.17 | <0.17 | <0.04 |
| eFG*(mL/min/1,73 m2) | 55 | 102 | 25 | 61 | 169 | >170 | >170 | >170 | |
| Albumin (g/dL) | 3.8-5.4 | 1.25 | 1.45 | 1.8 | 2.2 | 1.4 | 1.1 | 1.8 | 3.3 |
| Cholesterol/ triglycerides (mg/dL) | 60-190/35-165 | 286/322 | 280/420 | 285/335 | 194/286 | 245/290 | 297/491 | 229/244 | 172/150 |
| C3/C4 (mg/dL) | 90-150/15-35 | 39/15 | 49/15 | 85/30 | 97/25 | 82/19 | |||
| Leukocytes (mm3) | 6000-17500 | 31200 | 20890 | 16800 | 19100 | 17500 | 18850 | 30000 | 9240 |
| Haematocrit/ Haemoglobin (%/g/dL) | 37-39/10.5-13.5 | 26.4/8.4 | 20.3/6.9 | 25/8.3 | 32/10.9 | 29/9.3 | 30/10 | 34/11 | 37.5/11.2 |
| Platelets count (mm3) | 150000-450000 | 1436000 | 1058000 | 349000 | 679000 | 646000 | 675000 | 732000 | 204000 |
| LDH (UI/L)# | 200-764 | 563 | 382 | 567 | 356 | 580 | 442 | ||
| Proteinuria (g/L) | <0.15 | 13.3 | 9 | 6 | 3 | 2.6 | 0.12 | ||
| Urinary sediment | Abundant erythrocytes | 15 erythrocyte/field | >25 erythrocyte/field | 5-15 erythrocyte/field | >15 erythrocyte/field | 0-5 erythrocyte/field |
*Estimated glomerular filtration rate by Schwartz formula (K 0.45). #Lactate dehydrogenase.Day 6: Initiation of peritoneal dialysis. Day 14: 48 h after plasma infusion therapy. Day 39: First dose of eculizumab Day 46: 7 days after first dose of eculizumab. Day 65: 48 h after third dose of eculizumab. Day 270: 6 months after starting eculizumab
Based on clinical and laboratory findings a diagnosis of nephrotic syndrome of the first year of life was initially established while decreased C3 led us to investigate a potential infectious origin. However, blood, urine and stool cultures resulted negative. Likewise, viral stool studies and serological tests for syphilis, acquired immunodeficiency syndrome (AIDS), hepatitis B and C, cytomegalovirus, toxoplasmosis, parvovirus, mycoplasma and Epstein − Barr virus were all negatives. At that time treatment included albumin plus furosemide and amlodipine for blood pressure control; however, as urine output did not improve a renal biopsy was performed at day 4 after admission. The same day, first of 3 daily boluses of methylprednisolone (30 mg/kg/day) was given and peritoneal dialysis was started [Table 1]. The biopsy showed focal increase in mesangial cellularity, coinciding with reduction in capillary lumens, collapse of glomerular tufts and presence of microthrombi. We did not observe an increase in the mesangial matrix or double contours of the capillary walls employing silver stain. By immunofluorescence, focal and segmental deposits of IgM alone were seen. Also, focal luminal dilation in proximal convoluted tubules, disruption of the brush border and detached epithelial cells, mild interstitial chronic inflammatory infiltrates and isolated microthrombi at inter-tubular capillaries were observed. These findings supported the diagnosis of thrombotic microangiopathy [Figure 1]. On this basis, a diagnosis of TMA was made, which was unusual since there were no laboratory data consistent with mechanical hemolysis except for a fall in platelets counts >25% from baseline. A diagnosis of aHUS was established, and daily plasma infusions were administered as a bridge until starting eculizumab therapy. He was immediately vaccinated against Neisseria Meningitidis and Streptococcus Pneumoniae too. Of note, after two plasma infusions his urine output improved leading to discontinuation of dialysis [Table 1].
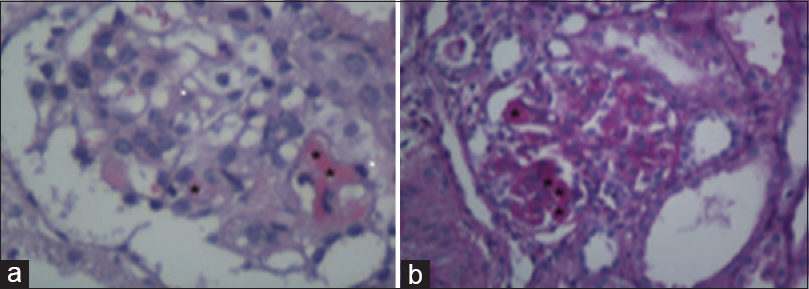
- (a) Representative image of a glomerulus with segmental eosinophilic intracapillary thrombi (black asterisks). Likewise, a slight segmental increase in mesangial cellularity was observed, due to ischemic retraction of the capillary loops. (Hematoxylin and Eosin, ×400); (b) In this case, intracapillary thrombi are clearly observed (black asterisks), linked to the collapse and retraction of the mesangial compartment. These changes are associate to the reduction of glomerular filtration rate. (Periodic Acid Schiff staining. ×400)
Simultaneously, the child was fully investigated. Accordingly, isolation of STEC, screening by polymerase chain reaction or detection of free Shiga toxin production in stools as well as lipopolysaccharides IgM antibodies for most common serotypes (O157, O145 and O121) resulted negative. ADAMTS 13 activity was normal (68%) and antibodies against ADAMTS 13 and CFH negative. Homocysteine level (6.2 μmol/l) was normal and antinuclear and anti-double stranded DNA antibodies were negative. Analysis of complement system corroborated a marked activation of the alternative pathway. C3 levels were persistently low (52 mg/dl) with normal C4 (15 mg/dl) concentration. Furthermore, serum functional activity of the classical pathway was 222 UH50/ml (normal value (NV) 180−280) but the alternative pathway was >60 min (NV 7−12). C3 nephritic factor and C3 degradation products (C3d) were negative. Factor B 197 mg/L (NV 229−394), factor H 409 mg/L (NV 329−557), factor I 18 mg/L (NV 15−31) and properdin 16 mg/dL (NV 19−50) levels were within normal ranges. Screening genes encoding complement regulatory proteins, coagulation and other genes causative of aHUS by next generation sequencing (NGS) excluded pathogenic mutations in the following genes: ADAMTS13, C3, CD46/MCP, CFB, CFH, CFHR1, CFHR2, CFHR3, CFHR5, CFI, DGKE, MMACHC, PIGA, PLG, and THBD. Further, deletion/duplication of CFH, CFHR1, CFHR2, CFHR3, CFHR5 genes were ruled out by multiplex ligation-dependent probe amplification (MLPA), with an overall diagnostic specificity >99.9%. However, a heterozygous variant in the CFHR3 gene (c.613+2T>C) of uncertain significance was detected. In the following days the patient persisted with edema, but blood pressure became normal without medication and also he recovered the renal function and normalized complement levels. On day 20, treatment was switched to eculizumab at a dose of 300 mg per week for 2 weeks and then every 3 weeks adjusted to patient weight (7.4 kg). At the time of starting eculizumab he had normal renal function but nephrosis persisted [Table 1]. Notably, after the second dose edema disappeared but hypoalbuminemia and massive proteinuria persisted. No adverse effects of eculizumab use were recorded. Because of familiar reasons the patient continued his follow-up in another country. After 6 months of treatment with eculizumab patient outcome was favorable, with normal renal function, remission of the nephrotic syndrome and without signs of TMA.
Discussion
Although aHUS is defined by the presence of thrombocytopenia (<150,000/mm3), microangiopathic hemolytic anemia and renal damage manifested by AKI and/or proteinuria and hematuria, 25% of the patients do not have all the three diagnostic criteria.[1] Some authors also consider thrombocytopenia criterion as a fall >25% in platelet count from baseline, as seen in our patient.[4] In addition, the low incidence of the disease and the heterogeneity of its clinical picture makes its diagnosis even more difficult. The disease can be triggered by multiple factors,[15] among them, vaccines against hepatitis B and influenza have been recognized.[67] Despite clinical onset in our patient after vaccination, it is uncertain whether this was a temporal association or the cause of activation of latent defects of complement system. Initial symptoms included diarrhea, which is seen in ~40% of patients at disease onset[2] and that highlight the need to exclude the possibility of STEC-HUS.[12] As mentioned, our patient presented with dehydration and after volume infusion he developed edema but, remarkably, hypoalbuminemia and dyslipidemia were detected in the very first laboratory test, prior to fluid administration. On this basis, we suspected a nephrotic syndrome of the first year of life with low C3 levels; thus a potential infectious cause was considered prior to perform a renal biopsy.[89]
The lack of hemolysis related findings in our patient is unclear. However, it has been noted that 15% of aHUS patients may have normal platelets count.[3] Our case had marked thrombocytosis at admission, which was assumed as acute-phase reactant or secondary to the nephrotic syndrome. However, as decrease platelet counts were observed, we retrospectively thought that TAM might had contributed to that finding. After biopsy proven diagnosis of TMA, the subtype of aHUS resulting from recessive mutations in the DGKE gene was suspected since those gene mutations can cause nephrotic syndrome during the first year of life due to associated membranoproliferative glomerulonephritis.[1310] However, biopsy findings suggesting membranoproliferative glomerulonephritis were not seen in our patient [Figure 1].[1112] Furthermore, the observed complement activation was also not compatible with DGKE nephropathy and more important, DGKE mutations were excluded by genetic analysis including next generation sequencing (NGS), with a diagnostic specificity >99.9%. Another possible explanation is that our patient had mutations of the thrombomodulin (THBD) gene, which may manifest as TMA with complement activation and also have been exceptionally reported as nephrotic syndrome at the beginning of the disease.[1314] Nevertheless, mutations of THBD gene were also not found. In contrast, we found a variant in the CFHR3 gen classified of uncertain significance (class 3) according to the American College of Medical Genetics. Although the CFHR3 gene variant in exon 4 (c.613+2T>C) found in our patient, was classified as a variant of uncertain significance, it is important to remember that the affected sequence is highly preserved. CFHR3 gene pathogenic mutations had been related with increased susceptibility for aHUS.[15] It should be noted that complement gene mutations are not found in around 40% of patients with aHUS,[1] but that does not preclude a different response to eculizumab, as has been demonstrated in our patient and the literature with remission of aHUS despite absence of identified gene anomalies.[2]
Although we are aware that eculizumab is the cornerstone of aHUS treatment and considering that plasmapheresis was unavailable in our center, we initially administered daily plasma infusions until getting eculizumab for this patient. With this treatment, he experienced a rapid improvement leading to discontinuation of dialysis after two plasma infusions; although such dramatic response is uncommon, a similar benefit has already been reported in children with aHUS.[1617] It would be inferred that this therapy could add some functional circulating complement regulators, because shortly afterwards he normalized C3 levels and renal function; however, nephrosis symptoms persisted.[1819] Regardless of the cause of aHUS, with or without a demonstrated mutation or autoantibody, eculizumab, an humanized monoclonal antibody that binds to human C5 with high affinity, blocking C5 cleavage to C5a and C5b, have been associated with better outcomes. Consistently, its administration induced the progressive remission of humoral parameters of nephrotic syndrome.[19] Indeed our patient normalized not only renal function and blood pressure in few days, but proteinuria some months later while receiving eculizumab.
In conclusion, we here reported a case of aHUS manifested with nephrotic syndrome. This uncommon presentation outlines the need for a heightened diagnosis suspicion of this already rare disease since early initiation of eculizumab therapy is known to improve its prognosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- An update for atypical haemolytic uraemic syndrome: Diagnosis and treatment. A consensus document. Nefrologia. 2015;35:421-47.
- [Google Scholar]
- Clinical and genetic predictors of atypical hemolytic uremic syndrome phenotype and outcome. Kidney Int. 2018;94:408-18.
- [Google Scholar]
- An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31:15-39.
- [Google Scholar]
- Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney disease: Improving global outcomes” (KDIGO) controversies conference. Kidney Int. 2017;91:539-51.
- [Google Scholar]
- Clinical characteristics and genetic backgrounds of Japanese patients with atypical hemolytic uremic síndrome. Clin Exp Nephrol. 2018;22:1088-99.
- [Google Scholar]
- Hepatitis B vaccine-associated atypical hemolytic uremic syndrome. Turk J Haematol. 2013;30:418-9.
- [Google Scholar]
- Influenza-associated thrombotic microangiopathies. Pediatr Nephrol. 2018;33:2009-25.
- [Google Scholar]
- The etiology of congenital nephrotic syndrome: Current status and challenges. World J Pediatr. 2016;12:149-58.
- [Google Scholar]
- Podocyte dysfunction in atypical haemolytic uremic syndrome. Nat Rev Nephrol. 2015;11:245-52.
- [Google Scholar]
- DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J Am Soc Nephrol. 2013;24:377-84.
- [Google Scholar]
- Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45:531-6.
- [Google Scholar]
- Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345-57.
- [Google Scholar]
- Nephrotic-range proteinuria and peripheral edema in a child: Not only idiopathic nephrotic syndrome. Case Rep Nephrol Dial. 2016;6:120-as7.
- [Google Scholar]
- Genetic Atypical Hemolytic-Uremic Syndrome. 2007. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; :1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1367/
- [Google Scholar]
- Efficacy of plasma therapy in atypical hemolytic uremic syndrome with complement factor H mutations. Pediatr Nephrol. 2008;23:1363-6.
- [Google Scholar]
- Successful plasma therapy for atypical hemolytic uremic syndrome caused by factor H deficiency owing to a novel mutation in the complement cofactor protein domain 15. Am J Kidney Dis. 2005;45:415-21.
- [Google Scholar]
- Complement and the atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2008;23:1957-72.
- [Google Scholar]
- Terminal complement inhibitor eculizumab in atypical hemolytic uremic syndrome. N Engl J Med. 2013;6,368:2169-81.
- [Google Scholar]




